|
Developed by 
|
Supported by 
|
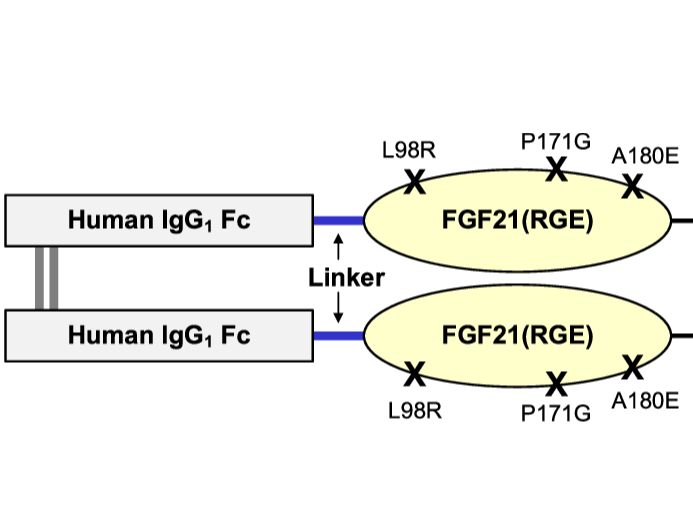
Efruxifermin
Developer(s)
|
|
Drug structure
Efruxifermin
Punegel et al. 2023
Drug information
Associated long-acting platforms
Monoclonal antibodies and antibody drug conjugates
Administration route
Subcutaneous
Therapeutic area(s)
Use case(s)
Use of drug
Ease of administration
User acceptance
Not provided
Dosage
Available dose and strength
Not provided
Frequency of administration
Not provided
Maximum dose
Not provided
Recommended dosing regimen
Not provided
Additional comments
Not provided
Dosage link(s)
Not provided
Drug information
Drug's link(s)
Generic name
Brand name
Compound type
Summary
Approval status
Regulatory authorities
Delivery device(s)
Not provided
Scale-up and manufacturing prospects
Scale-up prospects
Not provided
Tentative equipment list for manufacturing
Not provided
Manufacturing
Not provided
Specific analytical instrument required for characterization of formulation
Not provided
Clinical trials
AK-US-001-0106
Identifier
NCT06528314
Link
https://clinicaltrials.gov/study/NCT06528314
Phase
Phase III
Status
Recruiting
Sponsor
Akero Therapeutics, Inc
More details
This is a multi-center evaluation of efruxifermin (EFX) in a randomized, double-blind, placebo-controlled study in subjects with compensated cirrhosis due to NASH/MASH.
Purpose
A Study Evaluating Efruxifermin in Subjects With Compensated Cirrhosis Due to NASH/MASH
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2024-09-04
Anticipated Date of Last Follow-up
2025-05-23
Estimated Primary Completion Date
2029-09-01
Estimated Completion Date
2029-10-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
- Male
- Female
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Cohort 1: Biopsy proven compensated cirrhosis (fibrosis stage 4) due to NASH/MASH * Cohort 2: Biopsy proven or non-invasively diagnosed compensated cirrhosis (fibrosis stage 4) due to NASH/MASH Exclusion Criteria: * Other causes of liver disease based on medical history and/or liver histology and/or central laboratory results * Type 1 diabetes or unstable Type 2 diabetes * Any current or prior history of decompensated liver disease Other inclusion and exclusion criteria may apply
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
1150
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Primary outcome: Time from randomization to first occurrence of disease progression as measured by composite of protocol-specified clinical events (5 years)
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
AK-US-001-0105
Identifier
NCT06215716
Link
https://clinicaltrials.gov/study/NCT06215716
Phase
Phase III
Status
Recruiting
Sponsor
Akero Therapeutics, Inc
More details
This is a multi-center evaluation of efruxifermin (EFX) in a randomized, double-blind, placebo-controlled study in subjects with non-cirrhotic NASH/MASH and fibrosis stage 2 or 3 (F2 or F3). The study will enroll subjects in two cohorts for a total samples size of 1650 subjects.
Purpose
A Study Evaluating Efruxifermin in Subjects With Non-Cirrhotic Nonalcoholic Steatohepatitis (NASH)/Metabolic Dysfunction-Associated Steatohepatitis (MASH) and Fibrosis
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2023-12-01
Anticipated Date of Last Follow-up
2025-05-09
Estimated Primary Completion Date
2032-11-01
Estimated Completion Date
2032-11-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
- Male
- Female
Accepts pregnant individuals
No
Accepts lactating individuals
No
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Males and non-pregnant, non-lactating females between 18 - 80 years of age inclusive, based on the date of the screening visit. * Previous history or presence of 2 out of 4 components of metabolic syndrome (obesity, dyslipidemia, elevated blood pressure, elevated fasting glucose) or type 2 diabetes. * Cohort 1: Biopsy-proven NASH/MASH. Must have had a liver biopsy obtained ≤ 180 days prior to screening with fibrosis stage 2 or 3 and a non-alcoholic fatty liver disease (NAFLD) activity score (NAS) of ≥ 4 with at least a score of 1 in each of the following NAS components: * Steatosis (scored 0 to 3), * Ballooning degeneration (scored 0 to 2), and * Lobular inflammation (scored 0 to 3). Exclusion Criteria: * Other causes of liver disease based on medical histor
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
1650
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
AK-US-001-0107
Identifier
NCT06161571
Link
https://clinicaltrials.gov/study/NCT06161571
Phase
Phase III
Status
Enrolling by invitation
Sponsor
Akero Therapeutics, Inc
More details
The aim of this study is to assess the safety and tolerability of EFX compared to placebo in subjects with non-invasively diagnosed NASH/MASH and NAFLD/MASLD.
Purpose
A Study Evaluating Efruxifermin in Subjects With Non-invasively Diagnosed Nonalcoholic Steatohepatitis (NASH)/Metabolic Dysfunction-Associated Steatohepatitis (MASH) and Nonalcoholic Fatty Liver Disea
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2023-11-10
Anticipated Date of Last Follow-up
2025-01-31
Estimated Primary Completion Date
2026-04-01
Estimated Completion Date
2026-10-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
- Male
- Female
Accepts pregnant individuals
No
Accepts lactating individuals
No
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: Main Study Only: * Males and non-pregnant, non-lactating females between 18 - 80 (between 19-80 in the Republic of Korea) years of age inclusive, on the day of signing informed consent * Previous history or presence of 2 out of 4 components of metabolic syndrome (obesity, dyslipidemia, elevated blood pressure, elevated fasting glucose) or type 2 diabetes * Suspected or confirmed diagnosis of NASH/MASH or NAFLD/MASLD or non-invasively diagnosed NASH/MASH or NAFLD/MASLD Open-Label Rollover * Prior participation in the placebo arm of a previous Akero Phase 2 study Exclusion Criteria: * Other causes of liver disease based on medical history and/or liver histology and/or central laboratory results, including but not limited to: alcoholic liver disease, autoimmune disor
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
700
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
20100018
Identifier
NCT01856881
Link
https://clinicaltrials.gov/study/NCT01856881
Phase
Phase I
Status
Terminated
Sponsor
Amgen
More details
The purpose of this study is to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics following ascending multiple doses of AMG 876 in subjects with type 2 diabetes.
Purpose
Multiple Ascending Dose Study in Subjects With Type 2 Diabetes
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2013-03-01
Anticipated Date of Last Follow-up
2015-11-05
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2014-11-01
Actual Completion Date
2015-03-01
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Male and female subjects ≥ 18 to ≤ 65 years of age at the time of randomization * Female subjects must be of documented non-reproductive potential * Diagnosed with type 2 diabetes * HbA1c ≥ 6.5% and ≤ 10% * Fasting C-peptide value ≥ 0.8 ng/mL * Body mass index (BMI) between ≥ 25.0 and ≤ 40.0 kg/m2 at screening Exclusion Criteria: * Female subjects who are lactating/breastfeeding or who plan to breastfeed while on study through 4 weeks after receiving the last dose of study drug. * Male subjects with partners who are pregnant or planning to become pregnant while the subject is on study through 4 weeks after receiving the last dose of study drug * Evidence or history at screening of diabetic complications with significant end-organ damage, eg, proliferative retinopat
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
86
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Not provided
Studied route(s) of administration
Not provided
Use case
Not provided
Key resources
20100015
Identifier
NCT01492465
Link
https://clinicaltrials.gov/study/NCT01492465
Phase
Phase I
Status
Terminated
Sponsor
Amgen
More details
The purpose of this study is to determine whether AMG 876 is safe and well tolerated in subjects with type 2 diabetes.
Purpose
Single Ascending Dose Trial in Patients With Type 2 Diabetes
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2011-11-01
Anticipated Date of Last Follow-up
2013-02-13
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2012-07-01
Actual Completion Date
2012-10-01
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Subject has provided written informed consent * Men and women between the ages of 18 and 65, inclusive at the time of randomization * Women must be of documented non-reproductive potential (ie, postmenopausal \[see definition below\]; OR history of hysterectomy; OR history of bilateral tubal ligation; OR history of bilateral oophorectomy). * Diagnosed with type 2 diabetes * HbA1c ≥ 6.5% and ≤ 10% * Fasting C-peptide value ≥ 0.8 ng/mL * Men must agree for the duration of the study and continuing for 4 weeks after the dose of study drug, to practice a highly effective method of birth control. Highly effective methods of birth control include sexual abstinence, vasectomy or a condom with spermicide (men) in combination with either barrier methods, hormonal birth control
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
47
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Not provided
Studied route(s) of administration
Not provided
Use case
Not provided
Key resources
Harmony
Identifier
NCT04767529
Link
https://clinicaltrials.gov/study/NCT04767529
Phase
Phase II
Status
Completed
Sponsor
Akero Therapeutics, Inc
More details
This is a multi-center evaluation of efruxifermin (EFX) in a randomized, double-blind, placebo-controlled study in non-cirrhotic subjects with biopsy-proven F2 - F3 NASH.
Purpose
A Study of Efruxifermin in Non-Cirrhotic Subjects With Histologically Confirmed Nonalcoholic Steatohepatitis (NASH)
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2021-02-16
Anticipated Date of Last Follow-up
2025-04-23
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2024-05-02
Actual Completion Date
2024-05-02
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Males and non-pregnant, non-lactating females between 18 - 75 years of age inclusive, based on the date of the screening visit. * Previous history or presence of 2 out of 4 components of metabolic syndrome (obesity, dyslipidemia, elevated blood pressure, elevated fasting glucose) or type 2 diabetes. * FibroScan measurement \> 8.5 kPa \[kilopascal\]. * Biopsy-proven NASH. Must have had a liver biopsy obtained ≤ 180 days prior to randomization with fibrosis stage 2 to 3 and a non-alcoholic fatty liver disease (NAFLD) activity score (NAS) of ≥ 4 with at least a score of 1 in each of the following NAS components: * Steatosis (scored 0 to 3), * Ballooning degeneration (scored 0 to 2), and * Lobular inflammation (scored 0 to 3). Exclusion Criteria: * Weight loss \
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
128
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Not provided
Studied route(s) of administration
Not provided
Use case
Not provided
Key resources
Excipients
Proprietary excipients used
Not provided
Novel excipients or existing excipients at a concentration above Inactive Ingredients Database (IID) for the specified route of administration
Not provided
Residual solvents used
Not provided
Patent info
Description
Efruxifermin compositions
Brief description
Efruxifermin compositions, comprising sugar, arginine/arginine-HCI or arginine/glutamic acid, and a surfactant disclosed in different ratios and concentrations
Representative patent
WO2023064808
Category
Formulation
Patent holder
AKERO THERAPEUTICS, INC.
Exclusivity
Not provided
Expiration date
September 11, 2043
Status
Pending: AE, AP, AU, BR, CA, CL, CN, CO, CR, EA, EG, EP, HK, ID, IL, IN, JO, JP, KR, MA, MX, MY, NZ, PA, PE, PH, SA, SG, TH, UA, US, VN, ZA
Description
Method of Treatment using Efruxifermin
Brief description
Method of treating metabolic disorder (incl. type 1 diabetes, obesity etc.) using Efruxifermin (SEQ ID NO 41)
Representative patent
WO2013033452
Category
Method of treatment
Patent holder
AMGEN INC.
Exclusivity
Not provided
Expiration date
August 30, 2032
Status
Granted: AU Pending: JP, MX, US Not in force: EP
Description
Specific Efruxifermin polypeptides
Brief description
Efruxifermin polypetide sequence specifically claimed as SEQ ID NO 47
Representative patent
WO2010129503
Category
Compound
Patent holder
AMGEN INC.
Exclusivity
Not provided
Expiration date
May 4, 2030
Status
Granted: AR, BR, AU, CA, CN, CO, CR, EA (KZ, RU), EP (CH, DE, FR, IT, GB, LI), HK, IN, ID, IL, JP, KR, LB, MY, MX, MA, NZ, PA, GC, PE, PH, SG, ZA, TH, TW, TN, UA, US Pending: BW, EG, JO, LY, VN
Description
Broad Efruxifermin polypeptides
Brief description
Broad FGF21 mutant polypeptides, covering mutants of Efruxifermin IgG constant domain as SEQ ID NO 13 and Efruxifermin FGF21 as SEQ ID NO 4, and tris(tetraglycylseryl) peptide linker as SEQ ID NO 23 Full Efruxifermin sequence not disclosed
Representative patent
WO2009149171
Category
Compound
Patent holder
AMGEN INC.
Exclusivity
Not provided
Expiration date
June 3, 2029
Status
Granted: DZ, AU, BR, CA, CN, CO, EA (KZ, RU), EP (CH, DE, FR, GB, LI), HK, IN, ID, IL, JP, KR, MY, MX, MA, NZ, PE, SG, ZA, TW, TN, UA, US, VN Pending: AR, BW, CL, CR, EG, JO, LY, GC, UY
Supporting material
Publications
Shanaka Stanislaus, Randy Hecht, Junming Yie, Todd Hager, Michael Hall, Chris Spahr, Wei Wang, Jennifer Weiszmann, Yang Li, Liying Deng, Dwight Winters, Stephen Smith, Lei Zhou, Yuesheng Li, Murielle M. Véniant, Jing Xu, A Novel Fc-FGF21 With Improved Resistance to Proteolysis, Increased Affinity Toward β-Klotho, and Enhanced Efficacy in Mice and Cynomolgus Monkeys, Endocrinology, Volume 158, Issue 5, 1 May 2017, Pages 1314–1327, https://doi.org/10.1210/en.2016-1917
Fibroblast growth factor (FGF) 21 is a natural hormone that modulates glucose, lipid, and energy metabolism. Previously, we engineered an Fc fusion FGF21 variant with two mutations, Fc-FGF21(RG), to extend the half-life and reduce aggregation and in vivo degradation of FGF21. We now describe a new variant developed to reduce the extreme C-terminal degradation and improve the binding affinity to β-Klotho. We demonstrate, by introducing one additional mutation located at the C terminus of FGF21 (A180E), that the new molecule, Fc-FGF21(RGE), has gained many improved attributes. Compared with Fc-FGF21(RG), Fc-FGF21(RGE) has similar in vitro potency, preserves β-Klotho dependency, and maintains FGF receptor selectivity and cross-species reactivity. In vivo, Fc-FGF21(RGE) showed reduced susceptibility to extreme C-terminal degradation and increased plasma levels of the bioactive intact molecule. The circulating half-life of intact Fc-FGF21(RGE) increased twofold compared with that of Fc-FGF21(RG) in mice and cynomolgus monkeys. Additionally, Fc-FGF21(RGE) exhibited threefold to fivefold enhanced binding affinity to coreceptor β-Klotho across mouse, cynomolgus monkey, and human species. In obese and diabetic mouse and cynomolgus monkey models, Fc-FGF21(RGE) demonstrated greater efficacies to Fc-FGF21(RG), resulting in larger and more sustained improvements in multiple metabolic parameters. No increased immunogenicity was observed with Fc-FGF21(RGE). The superior biophysical, pharmacokinetic, and pharmacodynamic properties, as well as the positive metabolic effects across species, suggest that further clinical development of Fc-FGF21(RGE) as a metabolic therapy for diabetic and/or obese patients may be warranted.
Puengel, T., & Tacke, F. (2023). Efruxifermin, an investigational treatment for fibrotic or cirrhotic nonalcoholic steatohepatitis (NASH). Expert Opinion on Investigational Drugs, 32(6), 451–461. https://doi.org/10.1080/13543784.2023.2230115
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease and strongly associated with metabolic disorders: obesity, type 2 diabetes (T2D), cardiovascular disease. Persistent metabolic injury results in inflammatory processes leading to nonalcoholic steatohepatitis (NASH), liver fibrosis, and ultimately cirrhosis. To date, no pharmacologic agent is approved for the treatment of NASH. Fibroblast growth factor 21 (FGF21) agonism has been linked to beneficial metabolic effects ameliorating obesity, steatosis, and insulin resistance, supporting its potential as a therapeutic target in NAFLD.
Areas covered
Efruxifermin (EFX, also AKR-001 or AMG876) is an engineered Fc-FGF21 fusion protein with an optimized pharmacokinetic and pharmacodynamic profile, which is currently tested in several phase 2 clinical trials for the treatment of NASH, fibrosis and compensated liver cirrhosis. EFX improved metabolic disturbances including glycemic control, showed favorable safety and tolerability, and demonstrated antifibrotic efficacy according to FDA requirements for phase 3 trials.
Expert opinion
While some other FGF-21 agonists (e.g. pegbelfermin) are currently not further investigated, available evidence supports the development of EFX as a promising anti-NASH drug in fibrotic and cirrhotic populations. However, antifibrotic efficacy, long-term safety and benefits (i.e. cardiovascular risk, decompensation events, disease progression, liver transplantation, mortality) remain to be determined.
Harrison SA, Frias JP, Neff G, et al. Safety and efficacy of once-weekly efruxifermin versus placebo in non-alcoholic steatohepatitis (HARMONY): a multicentre, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol Hepatol. 2023;8(12):1080-1093. doi:10.1016/S2468-1253(23)00272-8
Background: Fibroblast growth factor 21 (FGF21) regulates metabolism and protects cells against stress. Efruxifermin is a bivalent Fc-FGF21 analogue that replicates FGF21 agonism of fibroblast growth factor receptor 1c, 2c, or 3c. The aim of this phase 2b study was to assess its efficacy and safety in patients with non-alcoholic steatohepatitis (NASH) and moderate (F2) or severe (F3) fibrosis.
Methods: HARMONY is a multicentre, randomised, double-blind, placebo-controlled, 96-week, phase 2b trial that was initiated at 41 clinics in the USA. Adults with biopsy-confirmed NASH, defined by a non-alcoholic fatty liver disease activity score (NAS) of 4 or higher and scores of 1 or higher in each of steatosis, ballooning, and lobular inflammation, with histological stage F2 or F3 fibrosis, were randomly assigned (1:1:1), via an interactive response system, to receive placebo or efruxifermin (28 mg or 50 mg), subcutaneously once weekly. Patients, investigators, pathologists, site staff, and the sponsor were masked to group assignments during the study. The primary endpoint was the proportion of patients with improvement in fibrosis of at least 1 stage and no worsening of NASH, based on analyses of baseline and week 24 biopsies (liver biopsy analysis set [LBAS]). A sensitivity analysis evaluated the endpoint in the full analysis set (FAS), for which patients with missing biopsies were considered non-responders. This trial is registered with ClinicalTrials.gov, NCT04767529, and is ongoing.
Findings: Between March 22, 2021, and Feb 7, 2022, 747 patients were assessed for eligibility and 128 patients (mean age 54·7 years [SD 10·4]; 79 [62%] female and 49 male [38%]; 118 [92%] white; and 56 [41%] Hispanic or Latino) were enrolled and randomly assigned to receive placebo (n=43), efruxifermin 28 mg (n=42; two randomised patients were not dosed because of an administrative error), or efruxifermin 50 mg (n=43). In the LBAS (n=113), eight (20%) of 41 patients in the placebo group had an improvement in fibrosis of at least 1 stage and no worsening of NASH by week 24 versus 15 (39%) of 38 patients in the efruxifermin 28 mg group (risk ratio [RR] 2·3 [95% CI 1·1-4·8]; p=0·025) and 14 (41%) of 34 patients in the efruxifermin 50 mg group (2·2 [1·0-5·0]; p=0·036). Based on the FAS (n=128), eight (19%) of 43 patients in the placebo group met this endpoint versus 15 (36%) of 42 in the efruxifermin 28 mg group (RR 2·2 [95% CI 1·0-4·8]; p=0·033) and 14 (33%) of 43 in the efruxifermin 50 mg group (1·9 [0·8-4·3]; p=0·123). The most frequent efruxifermin-related adverse events were diarrhoea (16 [40%] of 40 patients in the efruxifermin 28 mg group and 17 [40%] of 43 patients in efruxifermin 50 mg group vs eight [19%] of 43 patients in the placebo group; all events except one were grade 1-2) and nausea (11 [28%] patients in the efruxifermin 28 mg group and 18 [42%] patients in the efruxifermin 50 mg group vs ten [23%] patients in the placebo group; all grade 1-2). Five patients (two in the 28 mg group and three in the 50 mg group) discontinued due to adverse events. Serious adverse events occurred in four patients in the 50 mg group; one was defined as drug related (ulcerative esophagitis in a participant with a history of gastro-oesophageal reflux disease). No deaths occurred.
Interpretation: Efruxifermin improved liver fibrosis and resolved NASH over 24 weeks in patients with F2 or F3 fibrosis, with acceptable tolerability, supporting further assessment in phase 3 trials.
Additional documents
No documents were uploaded
Useful links
Access principles

|
Collaborate for developmentConsider on a case by case basis, collaborating on developing long acting products with potential significant public health impact, especially for low- and middle-income countries (LMICs), utilising the referred to long-acting technology Not provided |

|
Share technical information for match-making assessmentProvide necessary technical information to a potential partner, under confidentiality agreement, to enable preliminary assessment of whether specific medicines of public health importance in LMICs might be compatible with the referred to long-acting technology to achieve a public health benefit Not provided |

|
Work with MPP to expand access in LMICsIn the event that a product using the referred to long-acting technology is successfully developed, the technology IP holder(s) will work with the Medicines Patent Pool towards putting in place the most appropriate strategy for timely and affordable access in low and middle-income countries, including through licensing Not provided |
Comment & Information
Not provided