|
Developed by 
|
Supported by 
|
Buprenorphine
Developer(s)

|
Camurus AB Generic
https://www.camurus.com/
Sweden Camurus AB is a biopharmaceutical company that creates long-acting treatments for serious and chronic diseases. This company originated in 1991. Camurus, based on scientific understanding, aims to enhance patient outcomes through innovative therapies. They specialise in pharmaceutical commercialization and have a portfolio that includes clinical trial products as well as two FDA-approved drugs. |
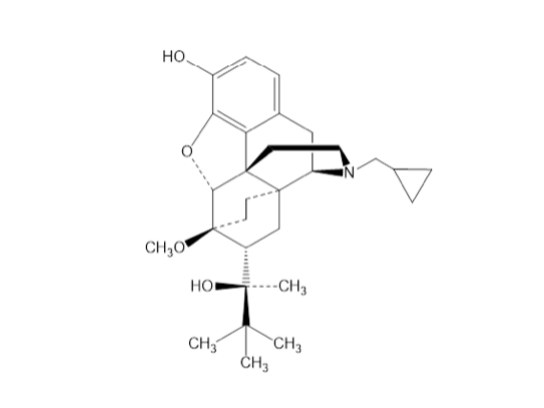
Drug structure
: (2S)-2-[17-(Cyclopropylmethyl)-4,5α-epoxy-3- hydroxy-6α,14-ethano-14α-morphinan-7α-yl]-3,3-dimethylbutan-2-ol.
https://www.brixadi.com/pdfs/brixadi-prescribing-information.pdf
Drug information
Associated long-acting platforms
FluidCrystal technology
Administration route
Subcutaneous
Therapeutic area(s)
Use case(s)
Use of drug
Ease of administration
Frequency of administration
User acceptance
Not provided
Dosage
Available dose and strength
• BRIXADI (weekly) is available in 8 mg/0.16 mL, 16 mg/0.32 mL, 24 mg/0.48 mL, and 32 mg/0.64 mL; • BRIXADI (monthly) is available in 64 mg/0.18 mL, 96 mg/0.27 mL, and 128 mg/0.36 mL prefilled syringe
Maximum dose
40mg/ week; 160 mg/ month
Recommended dosing regimen
1. The recommended weekly dose in patients not currently receiving buprenorphine treatment is 24 mg of BRIXADI (weekly) titrated up over the first week of treatment (administer a test dose of transmucosal buprenorphine 4 mg when objective signs of mild to moderate withdrawal appear). 2. If needed, during this first week of treatment, administer an additional 8 mg dose of BRIXADI (weekly), waiting at least 24 hours after the previous injection, for a total weekly dose of 32 mg BRIXADI (weekly). 3. Adults who have tolerated a single 4 mg dose of a transmucosal buprenorphine‐containing product. The test dose of transmucosal buprenorphine‐containing product should be administered based on instructions in the appropriate product label.
Additional comments
The recommended starting dose of Buvidal is 16 mg, with one or two additional 8 mg doses at least 1 day apart, to a target dose of 24 mg or 32 mg during the first treatment week. The recommended dose for the second treatment week is the total dose administered during the week of initiation. The maximum dose per week for patients who are on weekly Buvidal treatment is 32 mg with an additional 8 mg dose. The maximum dose per month for patients who are on monthly Buvidal treatment is 160 mg.


