|
Developed by 
|
Supported by 
|
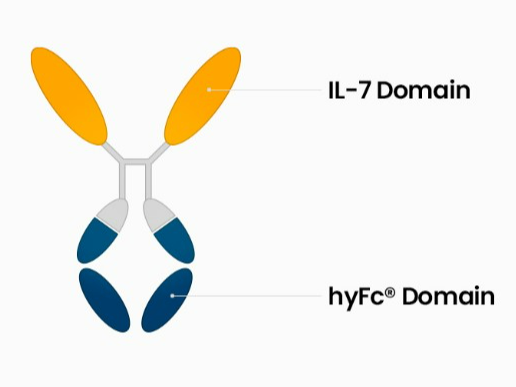
Long-acting hyFc Fusion Technology
Based on public informationDeveloper(s)

|
Genexine Originator
http://www.genexine.com/
South Korea Genexine is a leading South Korean biopharmaceutical company founded in 1999, focusing on developing and commercializing novel therapies using their two proprietary technologies for unmet medical needs. Their core R&D interest lies in immunotherapies for cancer and next-generation long-acting biologics. Their R&D efforts have yielded positive results. |
Sponsor(s)
|
No sponsor indicated |
Partnerships

|
|

|
Handok Pharmaceuticals, Inc. https://www.handok.co.kr |

|
I-MAB Biopharma https://www.i-mabbiopharma.com/ |

|
|

|
NeoimmuneTech https://neoimmunetech.com/ |

|
Hoffmann La Roche https://www.roche.com |

|
GenNBio, Inc. http://gennbio.com/ |

|
Turret Capital, Inc. https://www.turretfunds.com/ |

|
Ilkogen, Inc. https://www.ilkogen.com.tr/ |

|
CSPC Pharmaceutical Group ltd https://www.cspc.com.hk/en/global/home.php |

|
Fosun Pharma https://www.fosunpharma.com/en/ |

|
|

|
PharmaJet https://pharmajet.com/ |

|
Rezolute, Inc. https://www.rezolutebio.com/ |

|
Kingen Biotech www.kingenbiotech.com |

|
ToolGen, Inc. http://www.toolgen.com/eng |

|
EPD Biotherapeutics http://www.epdbio.com/home/en |
Technology information
Type of technology
Antibody Fragment proteins
Administration route
Intramuscular, Subcutaneous, Intravenous
Development state and regulatory approval
erythropoietin (EPO)
Marketed
Efesa is approved in Indonesia by the Indonesian Food and Drug Adminstration (BPOM)
Description
The novel noncytolytic hybrid Fc (hyFc) is a nonimmunogenic long-acting Fc fusion technology that uses national proteins to maximize drug stability. hyFc acts as a carrier of agonistic protein drugs using naturally existing IgD and IgG4 Fcs without any mutation in the hyFc region. IgD in the hyFC has high hinge flexibility, minimizing protein-to-protein interaction to increase drug efficacy, and lowering cytotoxicity problems through ADCC and CDC. By binding with neonatal Fc receptor (FcRn), IgG4 is recycled (regeneration) in vivo, enabling it to have long-acting pharmacokinetics.
Technology highlight
• Unmodified Natural Proteins as Carriers • Enhanced Efficacy with Reduced Cytotoxicity • Highly stable and versatile • Broad therapeutic potential • Potentially can be used in combination therapy
Technology main components
• Core Components: (i) Fc Fusion Protein: This protein is a hybrid of the Fc (fragment crystallizable) region of IgD and IgG4. (ii) IL-7 N-Terminals: These are two separate N-terminals (beginning sections) of the interleukin-7 protein bonded to the API and Fc fusion protein. • Additional Components: (I) Oligopeptide: This is a short chain of amino acids (II) Pharmaceutically accepted adjuvants -buffering agents, dispersing agents (for example: Sodium acetate, sodium chloride, potassium chloride, calcium chloride, Sodium lactate, etc)
Information on the raw materials sourcing, availability and anticipated price
Efesa (Long-acting erythropoietin) has been commercially marketed in Indonesia.
Delivery device(s)
No delivery device
APIs compatibility profile
API desired features
Proteins
Human growth hormone, bone morphogenetic protein-1 (BMP-1), growth hormone-releasing hormone, growth hormone-releasing peptide, interferons and interferon receptors (e.g., interferon-C, -3, and water-soluble type I interferon receptor, etc.), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), glucagon-like peptides (e.g., GLP-1), G-protein-coupled receptor, interleukins and interleukin receptors, enzymes, interleukin and cytokine binding proteins, macrophage activating factor, monoclonal/polyclonal antibodies are targeted.
Additional solubility data
Not provided
Additional stability data
Not provided
API loading: Maximum drug quantity to be loaded
75-90 wt%
API co-administration
2 different APIs : Therapeutic proteins can be combined however further information is not disclosed by the company
LogP
Not provided
Scale-up and manufacturing prospects
Scale-up prospects
Efesa (Efepoetin alfa), the inaugural hyFC product, has gained regulatory approval and is commercially accessible in Indonesia. Amid that, Genexine gained a 20,000 square-foot manufacturing facility located in Durham, North Carolina, United States.
Tentative equipment list for manufacturing
Not provided
Manufacturing
Manufacturing of Efesa (Efineptakin alfa) in a cleanroom involves: • Construction of a Plasmid Encoding IL 1 Ra-hyFc Fusion Protein • Establishment of Cell Lines Expressing the Present Fusion Protein • Confirmation of Protein Expression by Western • Blotting • Purification and Concentration of Protein • Characterization of hL1 RA-hFC Fusion Protein • Determination and Comparison of Binding Affinity • Detection of IL-8 Using ELISA
Specific analytical instrument required for characterization of formulation
• Field emission scanning electron microscope • Transmission electron microscopy • Photon correlation spectroscopy • Enzyme-linked immunosorbent assay (ELISA) • Circular dichroism (CD) spectroscopy • SDS-PAGE and Immunoblotting
Clinical trials
GX-I7-HV-001
Identifier
NCT02860715
Link
https://clinicaltrials.gov/study/NCT02860715
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
This is a Phase 1, randomized, double-blind, placebo-controlled, single ascending dose study designed to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of GX-I7 in healthy volunteers.
Purpose
Clinical Trial of GX-I7 in Healthy Volunteers
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2016-07-11
Anticipated Date of Last Follow-up
2018-10-21
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2017-01-20
Actual Completion Date
2018-06-02
Studied populations
Age Cohort
- Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: 1. Subject is willing and able to give informed consent after listening character of the clinical trial 2. Must be 19-45 years of age, inclusive 3. Weight 50-100kg, BMI 18-30kg/m2 4. Subject who is adequately able to attend the study based on medical history and physical exam, no clinically significant abnormality from vital sign and clinical laboratory values 5. No clinical abnormality from ECG test 6. Non-smoker (no smoking or no use of any product containing nicotine least for one month and negative from urine test) Exclusion Criteria: 1. Suspected or confirmed malignancy, or has malignancy history 2. Any clinically significant acute or chronic medical condition requiring care of a physician, in liver, biliary tract, renal, nervous system (CNS or peripheral).
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
30
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-G6_HV1
Identifier
NCT03651466
Link
https://clinicaltrials.gov/study/NCT03651466
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
This study is a single-center, double-blind, placebo-controlled, phase I study with healthy male subjects receiving ascending single s.c. doses of GX-G6
Purpose
Safety and Tolerability of GX-G6 in Healthy Male Subjects
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2017-08-31
Anticipated Date of Last Follow-up
2018-08-27
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2018-06-06
Actual Completion Date
2018-06-28
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria 1. male subjects aged between 18-50 years (both inclusive) 2. healthy subjects as determined by medical history, physical examination including vital signs, ECG and clinical laboratory testing 3. subjects who are able and willing to give written informed consent 4. male subjects must be using 2 acceptable methods for contraception (one of these methods should be a barrier method e.g. spermicide and condom) from start of dosing and refrain from fathering a child in the 3 months following dosing. Exclusion Criteria: History of: 1. clinically relevant allergy (except for untreated, asymptomatic, seasonal allergies at time of dosing), especially allergy to macrolide antibiotics; 2. any clinically significant pancreatic, hepatic, renal, gastrointestinal, cardiovascular.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
48
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-I7M-HPV-002
Identifier
NCT03144934
Link
https://clinicaltrials.gov/study/NCT03144934
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
This is a phase 1, randomized, double-blind, placebo-controlled, multiple ascending dose study to evaluate the safety and tolerability of GX-I7 in HPV-infected female volunteers
Purpose
Safety and Tolerability of GX-I7 in HPV-infected Female Volunteers
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2017-02-16
Anticipated Date of Last Follow-up
2018-10-21
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2018-03-22
Actual Completion Date
2018-03-22
Studied populations
Age Cohort
- Adults
Genders
- Female
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Subject willing and able to give informed consent * Must be ≥19 and ≤45 years diagnosed with HPV infection in two tests within screening periods or have history of HPV infection within 6 months and diagnosed with HPV infection in one test within screening periods * No clinical abnormality from ECG test * Must agree to use appropriate contraceptive methods (ie, condoms, cervical cap in conjunction with spermicide, sterilization, and intra uterine device) during the study and for 3 months after the last dose of study drug. Exclusion Criteria: * Subject with HSIL or more severe HPV infection * History of a known or suspected hypersensitivity, shock, or past history to the investigational drug or to similar drugs * Malignant tumor within 5 years other than successfully.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
32
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-H9-003
Identifier
NCT03309891
Link
https://clinicaltrials.gov/study/NCT03309891
Phase
Phase II
Status
Completed
Sponsor
Genexine, Inc.
More details
This is a randomized, open-label, active controlled, Phase 2 study designed to assess the safety, tolerability, efficacy, pharmacokinetics, and pharmacodynamics of weekly and semi-monthly doses of GX-H9 in the treatment of Paediatric Growth Hormone Deficiency (PGHD) as compared to the standard of care daily rhGH treatment.
Purpose
Dose Finding Study of GX-H9 in Paeditaric Patients With Growth Hormone Deficiency
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2016-01-18
Anticipated Date of Last Follow-up
2020-04-16
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2017-10-27
Actual Completion Date
2019-05-15
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Pre-pubertal children with either isolated GHD, or GH insufficiency as part of multiple pituitary hormone insufficiency, idiopathic or organic GH insufficiency (e.g., due to pituitary tumor, pituitary or brain surgery): * Boys: 3 years ≤ boy's age ≤ 11 years * Girls: 3 years ≤ girl's age ≤ 10 years 2. GHD confirmed by 2 different GH provocation tests with peak GH concentration below 10 ng/mL as described in consensus guidelines. Well documented historical GH provocation tests can be used for study eligibility providing that the tests are performed as defined in Appendix 2 (e.g. the same sampling time points). Data of each historical GH stimulation test will be reviewed by Medical Monitor and Sponsor in order to assess acceptance for the study.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
56
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Not provided
Studied route(s) of administration
Not provided
Use case
Not provided
Key resources
GX-I7-CA-010
Identifier
NCT05191784
Link
https://clinicaltrials.gov/study/NCT05191784
Phase
Phase II
Status
Not provided
Sponsor
Genexine, Inc.
More details
The purpose of this study is to evaluate the efficacy and safety of GX-I7 in combination with bevacizumab in subjects with recurrent glioblastoma.
Purpose
GX-I7 in Combination With Bevacizumab in Recurrent Glioblastoma (GBM) Patients
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2022-01-26
Anticipated Date of Last Follow-up
2023-06-08
Estimated Primary Completion Date
2024-12-31
Estimated Completion Date
2024-12-31
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Age ≥ 19 years 2. Histologically diagnosed glioblastoma patients who have been confirmed the progression of disease after attempting standard therapy (RT/CCRT and/or adjuvant chemotherapy (TMZ)) 3. Karnofsky Performance Status; KPS ≥ 60 or ECOG status 0-2 4. Life expectancy \> 12 weeks 5. Adequate hematologic and end organ function Exclusion Criteria: 1. Malignancies other than disease under study within 5 years prior to the first dose of study drug 2. Subjects who have received bevacizumab or other VEGF inhibitors prior to study participation 3. Body Mass Index (BMI) ≥ 30 kg/m2 4. Subjects confirmed intracranial hemorrhage with non-contrast CT or MRI 5. Clinically significant cardiovascular disease 6. History of arterial or venous thromboembolism 6 months prior t
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
20
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-G3
Identifier
NCT01951027
Link
https://clinicaltrials.gov/study/NCT01951027
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
pharmacokinetics/pharmacodynamics of GX-G3 after single subcutaneous administration in healthy male subjects.
Purpose
Phase I Study GX-G3 in Healthy Subjects
Interventions
Intervention 1
Intervention 2
Intervention 3
Intervention 4
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2013-09-01
Anticipated Date of Last Follow-up
2015-01-27
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2014-07-01
Actual Completion Date
2014-12-01
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: Subjects may be entered in the study only if they meet all of the following criteria: 1. Are capable of understanding and complying with the requirements of the study and have voluntarily signed the informed consent form (ICF); 2. Healthy male volunteers aged 20-45 years; 3. Have a body weight of 60-90 kg (inclusive), have a body mass index (BMI) equal to or greater than 19 and less than 27 kg/m2; 4. Are eligible for the study based on screening data (Subjects may participate if Investigator considered eligible after looking at other screening data); Exclusion Criteria: Subjects presenting with any of the following will not be entered in to the study: 1. Have a history of or current evidence of disease; 2. Have percent of white blood cell (WBC) or neutrophil \> UNL.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
53
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-P1-001
Identifier
NCT04298749
Link
https://clinicaltrials.gov/study/NCT04298749
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
This study is a single-center, double-blind, placebo-controlled, phase I study with healthy male volunteers receiving ascending single dose of GX-P1
Purpose
Safety and Tolerability of GX-P1 in Healthy Male Volunteers
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2020-08-11
Anticipated Date of Last Follow-up
2021-07-21
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2021-06-07
Actual Completion Date
2021-06-07
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: 1. Capable of understanding and complying with the requirements of the study and have voluntarily signed the informed consent form (ICF) 2. Healthy male volunteers aged 19-45 years within screening periods 3. Body weight of 50-90 kg, and body mass index (BMI) of 18.0-30.0 kg/m2 4. Healthy subjects as determined by medical history, physical examination vital signs, ECG and clinical laboratory testing Exclusion Criteria: 1. Any clinical significant pancreatic, hepatic, renal, gastrointestinal, cardiovascular, respiratory, hematological, central nervous system disease or other significant diseases which might influence either the safety of the subject or the absorption, metabolism or excretion of the active agent under investigation.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
24
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Not provided
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-G6-002
Identifier
NCT03962010
Link
https://clinicaltrials.gov/study/NCT03962010
Phase
Phase II
Status
Unknown status
Sponsor
Genexine, Inc.
More details
GX-G6-002 is a Phase 2, 12-week, randomized, parallel group, multi-centre, double blind, placebo-controlled and an open-label active comparator study.
Purpose
A Phase 2, 12-Week, Double-Blind, Efficacy and Safety of GX-G6 in Patients With Uncontrolled Type 2 Diabetes Mellitus
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
2019-06-01
Actual Start Date
Not provided
Anticipated Date of Last Follow-up
2019-05-22
Estimated Primary Completion Date
2022-06-01
Estimated Completion Date
2022-07-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Diagnosis of T2DM ≥ 6 months prior to screening 2. HbA1c level of 7-10% (inclusive) Exclusion Criteria: 1. Have known type 1 diabetes mellitus (T1DM) 2. History of severe hypoglycaemia defined as ≥ 2 episodes of severe hypoglycaemia within 6 months prior to screening 3. Have had ≥ 2 episodes of ketoacidosis or hyperosmolar state/coma requiring hospitalization in the 6 months prior to screening 4. Has fasting serum TG ≥ 500 mg/dL or 9 mmol/L at screening. 5. Patients receiving lipid-lowering therapy must have been on the same dose of therapy for the past three months
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
78
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX30_P1/2
Identifier
NCT03276988
Link
https://clinicaltrials.gov/study/NCT03276988
Phase
Phase I/II
Status
Completed
Sponsor
Genexine, Inc.
More details
This study is designed as a combination of phase 1 (Part A) and phase 2 (Part B). The purpose of Part A was to determine the safety, tolerability, and pharmacokinetics in patients with total thyroidectomy or near total thyroidectomy of GX-30 and it has been completed. The Part B is currently recruiting and will investigate the efficacy and safety of GX-30 compared with THYROGEN®.
Purpose
Tolerability, Safety and Pharmacokinetics Study of GX-30 in Total Thyroidectomy or Near Total Thyroidectomy Patients
Interventions
Intervention 1
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2017-09-07
Anticipated Date of Last Follow-up
2020-09-07
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2020-01-20
Actual Completion Date
2020-01-20
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Subjects who voluntarily consented, after listing enough explanation for this study and investigational product. * Minimum 19 years old. * Minimum 50kg of body weight. * Patients who had undergone total thyroidectomy or near total thyroidectomy due to differentiated thyroid carcinoma. * Patient undergoing thyroid hormone administration. Exclusion Criteria: * Thyroid cancer excluding differentiated thyroid carcinoma. * Thyroidectomy excluding total thyroidectomy and near total thyroidectomy. * Patients with heart, renal, or liver failure. * Patients with ischemic stroke or the history of ischemic stroke. * Smoker or Ex-smoker with less than 3 months of stopping * Patients with migraine or the history of migraine. * Patients that the researchers do not think fit into.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
8
Allocation
Randomized
Intervention model
Cross-over assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-H9-002
Identifier
NCT02946606
Link
https://clinicaltrials.gov/study/NCT02946606
Phase
Phase II
Status
Completed
Sponsor
Genexine, Inc.
More details
This is a randomized, active-controlled, open-label, sequential dose group, Phase 1b/2 study designed to assess the safety, tolerability, efficacy, pharmacokinetics, and pharmacodynamics of weekly and every other week doses of GX-H9 in the treatment of AGHD.
Purpose
A Clinical Study in AGHD to Assess Safety, Tolerability and Efficacy of GX-H9
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2015-01-01
Anticipated Date of Last Follow-up
2017-09-04
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2016-12-30
Actual Completion Date
2016-12-30
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria Each subject must meet all of the following criteria to be enrolled in this study: 1. Is a male or female aged ≥20 and 65 years with AGHD, either adult onset GHD due to hypothalamic pituitary disease or childhood onset GHD that is either idiopathic or due to hypothalamic pituitary disease or due to genetic causes. 2. Has documented confirmation (medical history) of GH deficiency during adulthood by 1 or more growth hormone (GH) stimulation tests, as follows: 3. Has been treated with stable hormonal replacement therapies for deficiencies of other hypothalamo pituitary axes and must have been on an optimized and stable treatment regimen for at least 3.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
45
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-E2_P2
Identifier
NCT02044653
Link
https://clinicaltrials.gov/study/NCT02044653
Phase
Phase II
Status
Completed
Sponsor
Genexine, Inc.
More details
The primary objective of study is * Part A : To explore the optimal fixed starting dose and dosing interval of GX-E2 * Part B : To evaluate the proof of concept (POC) of GX-E2
Purpose
Study to Evaluate the Efficacy and Safety of GX-E2 in the Anemic Patients Diagnosed With Chronic Kidney Disease (CKD)
Interventions
Intervention 1
Intervention 2
Intervention 3
Intervention 4
Intervention 5
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2014-04-15
Anticipated Date of Last Follow-up
2017-10-13
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2017-04-20
Actual Completion Date
2017-04-20
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Written informed consent * ≥18 yr of age * Chronic Kidney diseases with hemodialysis, peritoneal dialysis with Kt/V ≥ 1.2 (hemodialysis) or Kt/V ≥ 1.7 (peritoneal dialysis) within a year * Adequate transferrin saturation (≥20%), serum ferritin (≥100ug/L) * Should have received Vitamine B12 ≥ 3 months before the first dose of study agent * Should have received Folate ≥3 months before the first dose of study agent * No erythropoietin (EPO) therapy within 2 months before the planned first dose of GX-E2 and Hb\<10g/dL or No EPO therapy within a month (peritoneal dialysis) or 2 weeks (hemodialysis) before the planned first dose of GX-E2 and Hb\<10g/dL.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
257
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Single blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-E4-HV-003
Identifier
NCT06490939
Link
https://clinicaltrials.gov/study/NCT06490939
Phase
Phase I
Status
Not yet recruiting
Sponsor
Genexine, Inc.
More details
An open-label, parallel-group, single-center, Phase I study to compare the pharmacokinetic/pharmacodynamic characteristics, safety, and tolerability of a single intravenous administration of Efepoetin Alfa in healthy subjects
Purpose
Clinical Trial of Efepoetin Alfa in Healthy Subjects
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
2024-08-01
Actual Start Date
Not provided
Anticipated Date of Last Follow-up
2024-07-08
Estimated Primary Completion Date
2025-01-31
Estimated Completion Date
2025-08-31
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Key Inclusion Criteria: 1. Adult males and females between the ages of 19-45 2. Asian or Caucasian 3. Body weight \>50 kg and \<90 kg, BMI 18 \~30 (BMI(kg/m2) = Weight(kg) / {Height(m)}2) 4. Normal hemoglobin range. 5. Normal Serum ferritin and transferrin saturation range. 6. Normal serum folate range 7. Normal vitamin B12 range 8. White blood cell \>=3.0 X 10\^3 /mm3 9. Platelet \>= 150 X 10\^3/mm\^3 and \<450 X 10\^3/mm\^3 10. Nonsmoker or smoker who smokes below 10 cigarettes a day. Key Exclusion Criteria: 1. An allergy history, including drug allergies(example: aspirin, antibiotics, etc.) or clinically significant allergy. 2. Liver(including viral hepatitis), renal, respiratory, endocrine, neurological, immunological, blood, psychological, or circulatory system abnormalities, or a
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
40
Allocation
Not provided
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GX-I7-COV-009
Identifier
NCT04730427
Link
https://clinicaltrials.gov/study/NCT04730427
Phase
Phase I
Status
Terminated
Sponsor
Genexine, Inc.
More details
This study is a phase 1b clinical trial to investigate the safety and preliminary effects of a single dose of a test drug or placebo to the subjects who has diagnosed as COVID-19 infection.
Purpose
Safety and Preliminary Efficacy Study of GX-I7 in Patients With COVID-19
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2021-03-24
Anticipated Date of Last Follow-up
2022-11-25
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2022-05-08
Actual Completion Date
2022-07-07
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Key Inclusion Criteria: 1. Subjects who have been confirmed to be COVID-19 corresponding to mild cases of severity categorization classified by FDA through polymerase chain reaction (PCR) test or virus gene test (sequencing) and who can be available to be administered within seven days from the date of manifestation. 2. Subjects who are or will be inpatient. Key Exclusion Criteria: 1. Patients with symptoms of moderate or higher in the severity classification presented by FDA have evidence of lower respiratory tract infection in their imaging findings or need supplemental oxygen therapy or mechanical respiration (ie, non-invasive ventilation, invasive mechanical ventilation, extracorporeal membrane oxygenation, etc) 2. Subjects with infectious diseases such as bacteremia or severe pneum
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
10
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Single blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-I7-CA-007
Identifier
NCT04065087
Link
https://clinicaltrials.gov/study/NCT04065087
Phase
Phase I/II
Status
Withdrawn
Sponsor
Genexine, Inc.
More details
This is a phase 1/2, randomized, placebo-controlled study to evaluate safety, tolerability, anti-tumor activity and impact on absolute lymphocyte count of GX-I7 plus adjuvant temozolomide combination regimen in patients with newly diagnosed with glioblastoma who completed standard concurrent chemo-radiation therapy (CCRT)
Purpose
Efficacy and Safety Study of GX-I7 Plus Adjuvant Temozolomide Combination in Patients With Newly Diagnosed Glioblastoma
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
2019-08-22
Actual Start Date
Not provided
Anticipated Date of Last Follow-up
2022-03-02
Estimated Primary Completion Date
2022-05-27
Estimated Completion Date
2022-08-27
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Signed Informed Consent Form (ICF) 2. Age ≥ 19 years 3. Gross total resection equal to or greater than 80% based on post-op MRI, compared to pre-op MRI (Patients requiring biopsy only is not eligible) 4. Patients newly diagnosed with glioblastoma either by imaging or pathology testing, requiring concurrent chemo-radiotherapy (CCRT) and adjuvant temozolomide chemotherapy with curative intent 5. Karnofsky score ≥ 60 6. Life expectancy \> 12 weeks Exclusion Criteria: 1. Gliomatosis cerebri 2. Isocitrate dehydrogenase 1 \& 2 mutation 3. Pregnant or breast feeding women
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
Not provided
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-I7-CA-005
Identifier
NCT03733587
Link
https://clinicaltrials.gov/study/NCT03733587
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
This is a Phase 1b study to explore the safety and efficacy of GX-I7 in combination with CPA in patients with metastatic or recurrent solid tumors.
Purpose
GX-I7 With Cyclophosphamide in Patients With Metastatic or Recurrent Solid Tumors
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2018-10-17
Anticipated Date of Last Follow-up
2020-05-12
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2020-04-02
Actual Completion Date
2020-05-13
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Histological confirmation of solid tumor, for which no standard therapy exists or is available any longer. * Aged ≥19 years(Korean age). * Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. * Life expectancy ≥12 weeks. * Adequate hematological and end organ function defined by the following * laboratory results obtained within 14 days prior to Cycle 1 Day 1 (C1D1) * Female subjects of childbearing potential (including a female who has undergone tubal ligation) requires a negative serum pregnancy test performed within 14 days prior to C1D1. * Have an evaluable lesion(s) by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 * Subjects to be enrolled in the Phase 2a stage must be able to provide pre-treatment tissue biopsy or archival tissue
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
24
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GBM
Identifier
NCT03619239
Link
https://clinicaltrials.gov/study/NCT03619239
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
Patients will be enrolled in two stages: * Dose-escalation stage: Approximately 12-24 patients will be enrolled.
Purpose
Dose-escalation Study to Evaluate the Safety and Tolerability of GX-I7 in Patients With Glioblastoma
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2018-06-20
Anticipated Date of Last Follow-up
2020-11-08
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2020-09-25
Actual Completion Date
2020-09-25
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria 1. Ability to understand and willingness to sign a written informed consent document (ICF). 2. Age ≥ 19 years 3. Able to comply with the study protocol, in the investigator's judgment 4. Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Exclusion Criteria 1. Unable to comply with study and follow-up procedures 2. Is pregnant or breastfeeding 3. Have clinically significant cardiac disease (New York Heart Association, Class II or greater) including myocardial infarction, unstable arrhythmias, and/or unstable angina in the past 3 months 4. Have clinically significant liver disease, including alcoholic, or other hepatitis, cirrhosis, and inherited liver disease or current alcohol abuse.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
15
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-I7-CA-003
Identifier
NCT03478995
Link
https://clinicaltrials.gov/study/NCT03478995
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
Patients will be enrolled in two stages: * Dose-escalation stage: Approximately 15-30 patients will be enrolled. * Dose-expansion stage: 6-12 patients will be enrolled. Dose-escalation slots will be filled first, then dose-expansion slots.
Purpose
Study to Evaluate Safety and Tolerability of GX-I7 in Patients With Locally Advanced or Metastatic Solid Tumors
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2018-03-05
Anticipated Date of Last Follow-up
2020-05-07
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2020-03-16
Actual Completion Date
2020-03-16
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Signed Informed Consent Form (ICF) * Age ≥ 19 years * Able to comply with the study protocol, in the investigator's judgment * Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 * Life expectancy ≥ 12 weeks * Adequate hematologic and end organ function, defined by the following laboratory results obtained within 14 days prior to the first study treatment (Cycle 1, Day 1) * Serum pregnancy test for women of childbearing potential (including women who have had a tubal ligation) must be performed and documented as negative within 14 days prior to Cycle 1, Day 1 * For men: agreement to remain abstinent (refrain from heterosexual intercourse) or use contraceptive measures, and agreement to refrain from donating sperm
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
35
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-E2-P1
Identifier
NCT02291991
Link
https://clinicaltrials.gov/study/NCT02291991
Phase
Phase I
Status
Completed
Sponsor
Genexine, Inc.
More details
This is a randomized, placebo controlled, single dose study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of GX-E2 in healthy male subjects.
Purpose
Study to Evaluate Safety, Tolerability, and Pharmacokinetics/Pharmacodynamics of GX-E2 in Healthy Subjects
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2014-11-01
Anticipated Date of Last Follow-up
2015-01-27
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2015-01-01
Actual Completion Date
2015-01-01
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: * Written informed consent * Male subjects 20 to 55 years old * Adequate body weigth and BMI(19 ≤ BMI ≤ 27, 60.0kg ≤ body weigth ≤ 90.0kg) * The subject doesn't have a clinically significant abnormal laboratory value and/or clinically significant unstable medical or disease history. * Are eligible for the study hemoglobin data(12.0g/dL ≤ Hb ≤ 16.5g/dL) (Data is checked per 2 weeks within 28 days) * Adequate transferrin saturation, serum ferritin within 28 days * Adequate folate within 28 days * Adequate vitamin B12 within 28 days * Adequate WBC count (≥ 3.0 X 1000 µL) * Adequate PLT count(≥ 140 X 1000 µL) * nonsmoker or smoker smoked under 10 cigarettes a day Exclusion Criteria: * The subject has a clinically significant abnormal allergy including medical allergy.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
10
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
GXI7KGBio-001
Identifier
NCT04810637
Link
https://clinicaltrials.gov/study/NCT04810637
Phase
Phase II
Status
Unknown status
Sponsor
PT Kalbe Genexine Biologics
More details
This is a Phase 2 prospective, randomized, placebo-controlled, double-blinded, parallel group, single administration, multi-center study to assess the safety and efficacy of efineptakin alfa single treatment compared to placebo in elderly participants (adults ≥50years) with asymptomatic or mild COVID-19
Purpose
A Study to Evaluate the Safety and Efficacy of GX-I7 in Elderly Patients With Asymptomatic or Mild Symptoms of COVID-19
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2020-11-01
Anticipated Date of Last Follow-up
2021-03-22
Estimated Primary Completion Date
2021-06-01
Estimated Completion Date
2021-09-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Adults aged 50 years and above at the time of consent 2. Subjects who have been confirmed to be COVID-19 corresponding to asymptomatic case or mild cases of severity categorization classified by FDA through authorized molecular saliva-based test or polymerase chain reaction (PCR) test and who can be available to be administered within 7 days from the onset of any symptoms. 3. Patients who provide a voluntarily consent to participate in the study and sign the consent form in his/her own handwriting. 4. Female patients of childbearing potential (including female received a tubal ligation) should be prove negative pregnancy through pregnancy test before 24 hours of the IP administration, and must be willing to maintain abstinence (restraint sexual relationships).
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
210
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Triple-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
GX-I7-CA-006
Identifier
NCT03752723
Link
https://clinicaltrials.gov/study/NCT03752723
Phase
Phase I/II
Status
Completed
Sponsor
Genexine, Inc.
More details
To evaluate the safety and tolerability of escalating doses GX-I7 in combination with standard dose pembrolizumab, and to evaluate objective response rate (ORR) in subjects with refractory or relapsed TNBC
Purpose
Study of GX-I7 in Combination With Pembrolizumab in Refractory or Relapsed (R/R) TNBC Subjects(GX-I7-CA-006/KEYNOTE-899)
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2019-03-27
Anticipated Date of Last Follow-up
2024-03-01
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2023-05-11
Actual Completion Date
2023-05-11
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- Female
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Key Inclusion Criteria: 1. Triple negative must be defined as guidelines of American Society of Clinical Oncology(ASCO)/ College of American Pathologist(CAP): Estrogen Receptor (ER) \< 1% positive tumor nuclei, Progesterone Receptor (PR) \< 1% positive tumor nuclei, and negative for HER2 by IHC 1+, 0 or negativity status confirmed by in situ hybridization (ISH). 2. Subject must have received anthracycline and taxane based chemotherapy for TNBC 3. Has measurable disease as defined by RECIST 1.1 as assessed by the the local site Investigator/radiology. 4. Female subjects, age ≥ 19 years at the time of consent. Key Exclusion Criteria: 1. Known severe hypersensitivity (≥ Grade 3) to pembrolizumab, pembrolizumab formulation excipients or GX-I7 formulation excipients or cyclophosphamide.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
84
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Studied route(s) of administration
Not provided
Use case
Treatment
Key resources
Excipients
Proprietary excipients used
No proprietary excipient used
Novel excipients or existing excipients at a concentration above Inactive Ingredients Database (IID) for the specified route of administration
No novel excipient or existing excipient used
Residual solvents used
No residual solvent used
Additional features
Other features of the technology
- Single-use
- Other(s)
Drug release from the protein fragments at intercellular level
Release properties
The fusion protein GX-H9, comprising the erythropoietin analog -Efepoietin Alfa and the Fc domain of human IgG4, exhibits a prolonged serum half-life due to FcRn-mediated endocytosis and recycling. The gradual release of Efepoietin Alfa from the IgG4 moiety contributes to this extended pharmacokinetic profile. Following subcutaneous administration, mean serum concentrations of Efepoietin Alfa reached peak levels within 0-12 hours and subsequently declined in a biphasic pattern.
Injectability
HyFC prefilled injections are administered via subcutaneous or intramuscular injection. The needle is inserted into the designated site, and the medication is injected at a controlled rate
Safety
Clinical studies of Efesa Q2W show that 69.7% of the treated anemia in CKD-non-dialysis patients have experienced an AE (vs 64.5%).
Stability
hyFC technology products may allow long-term storage at cold storage with a shelf life of 1 year.
Storage conditions and cold-chain related features
Store in a refrigerator at temperatures between 2°C and 8°C. Avoid freezing. Protect from light and handle gently.
Potential application(s)
Therapeutic area(s)
Use case(s)
Use of technology
Ease of administration
- Administered by a community health worker
- Administered by a nurse
- Administered by a specialty health worker
Frequency of administration
Monthly, Every two weeks and Twice monthly
User acceptance
Not provided
Targeted user groups
Age Cohort- Children
- Adolescents
- Adults
- Older Adults
- All
Pregnant individuals
No
Lactating individuals
No
Healthy individuals
Unspecified
Comment
Not provided
Potential associated API(s)
Interleukins
Class(es)
Antineoplastic agent
Development stage
Phase II
Clinical trial number(s)
NCT03144934
Foreseen/approved indication(s)
Lymphopenia, solid tumors, infectious disease
Foreseen user group
Not provided
Foreseen duration between application(s)
Twice monthly
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
erythropoietin (EPO)
Class(es)
Erythropoiesis-stimulating agent
Development stage
Marketed
Clinical trial number(s)
NCT06466785
Foreseen/approved indication(s)
CKG induced Anemia
Foreseen user group
Chronic kidney diseases patients
Foreseen duration between application(s)
Every 2 weeks and 4 weeks
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Efesa is approved in Indonesia by the Indonesian Food and Drug Adminstration (BPOM)
Human Growth Hormone Agonists
Class(es)
Pituitary Hormones and analogues
Development stage
Phase III
Clinical trial number(s)
Not provided
Foreseen/approved indication(s)
Pediatric & Adult growth hormone deficiency
Foreseen user group
Children, adolescents and adults with Growth hormone deficiency
Foreseen duration between application(s)
Twice monthly
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Eftansomatropin alfa is currently in Phase 3 clinical study in China
Programmed cell death ligand 1 (PD-L1)
Class(es)
PDL1 inhibitors(Programmed death-ligand 1 inhibitors)
Development stage
Phase I
Clinical trial number(s)
NCT04298749
Foreseen/approved indication(s)
Autoimmune disease, and Organ Transplatation
Foreseen user group
Not provided
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Colony stimulating factors
Class(es)
Granulocyte colony-stimulating factor
Development stage
Phase I
Clinical trial number(s)
NCT01951027
Foreseen/approved indication(s)
Neutropenia
Foreseen user group
Not provided
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Glucagon-like peptide-1 (GLP-1) analogues (GLP-1)
Class(es)
GLP-1 and its analogues
Development stage
Phase I
Clinical trial number(s)
NCT03651466
Foreseen/approved indication(s)
Diabetes Mellitus and Obesity
Foreseen user group
Not provided
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Recombinant human thyroid-stimulating hormone (rhTSH)
Class(es)
Recombinant human thyroid stimulating hormone
Development stage
Phase I
Clinical trial number(s)
NCT03276988
Foreseen/approved indication(s)
Differentiated thyroid carcinoma
Foreseen user group
Patients who are undergoing Total or Partial Thyroidectomy
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
glucagon-like peptide-2 (GLP-2)
Class(es)
Glucagon-like peptide-2 (GLP-2) analogs
Development stage
Phase I
Clinical trial number(s)
Not provided
Foreseen/approved indication(s)
Short Bowel Syndrome
Foreseen user group
Not provided
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Interleukins
Class(es)
Interleukin-7
Development stage
Phase II
Clinical trial number(s)
NCT03478995
Foreseen/approved indication(s)
Fibrotic/metastatic cancers
Foreseen user group
Not provided
Foreseen duration between application(s)
Every three or six weeks
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Vascular Endothelial Growth Factor inhibitors (VEGF/VEGFR)
Class(es)
VEGR inhibitors
Development stage
Phase I
Clinical trial number(s)
Not provided
Foreseen/approved indication(s)
Neovascular (Wet) Age-Related Macular Degeneration
Foreseen user group
Not provided
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Programmed cell death ligand 1 (PD-L1)
Class(es)
PD-L1 inhibitors
Development stage
Phase I
Clinical trial number(s)
Not provided
Foreseen/approved indication(s)
Autoimmune disease and Organ Transplantion
Foreseen user group
Not provided
Foreseen duration between application(s)
Not provided
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Patent info
Description
Method for producing long-acting EPO-hybrid Fc fusion protein
Brief description
The present invention relates to a method for producing an Fc fusion protein comprising an EPO-hybrid Fc fusion protein (i.e., epoetin alpha) having a C-terminal lysine content of about 1–10% and a sialic acid content of about 17–27 mol/mol. The method comprises culturing host cells transformed with DNA encoding the Fc fusion protein and DNA encoding human α-2,3-sialyltransferase under optimized culture conditions, and purifying the Fc fusion protein using an optimized purification process including anion exchange chromatography.
Representative patent
WO2025221083
Category
Process
Patent holder
PT KALBE GENEXINE BIOLOGICS; GENEXINE, INC.
Exclusivity
Not provided
Expiration date
April 18, 2045
Status
Not yet in National Phase, deadline for entry 19 October 2026
Description
Modified IL-7 fusion protein formulations
Brief description
Provided is a pharmaceutical formulation comprising a modified IL-7 protein. More particularly, it comprises (a) a modified IL-7 fusion protein; (b) a basal buffer with a concentration of 10 to 50 mM; (c) a sugar with a concentration of 2.5 to 5 w/v %; and (d) a surfactant with a concentration of 0.05 to 6 w/v %. Such pharmaceutical formulation of a modified IL-7 fusion protein does not show aggregates formation, but shows protective effects on proteins under stress conditions such as oxidation or agitation, and thus can effectively be used for the treatment of a patient.
Representative patent
WO2017078385
Category
Formulation
Patent holder
Genexine Inc
Exclusivity
Not provided
Expiration date
November 2, 2036
Status
Granted: US, KR Pending: CN
Description
Composition comprising long-acting erythropoietin
Brief description
The present invention provides a composition containing long-acting erythropoietin, and a method for preparation thereof. More particularly, there is provided an EPO-Fc fusion protein composition with excellent bio-sustainability and high purity, wherein sialic acid content of EPO-Fc is 17 mol/mol or more, and host cell-derived protein (HCP) impurity is included in an amount of 100 ng/mg or less.
Representative patent
WO2017061780
Category
Formulation
Patent holder
GENEXINE, INC. GREEN CROSS CORPORATION
Exclusivity
Not provided
Expiration date
October 6, 2036
Status
Granted: AU, KR Pending: CN
Description
Modified IL-7 linked to oligopeptide consisting of 1-10aa
Brief description
The present invention provides a modified interleukin-7 and a use thereof. The modified IL-7 or an IL-7 fusion protein of the present invention comprising the same can be obtained in high yield, and biologically active in viral infection and cancer models. Therefore, they can be used for the prevention and treatment of various diseases.
Representative patent
WO2016200219
Category
Compound
Patent holder
Genexine Inc
Exclusivity
Not provided
Expiration date
June 10, 2036
Status
Granted: CA, CN, IN, JP, KR, US, RU Pending: EP, ID, HK Not in force: BR
Description
Method of treatment of anemia using fusion polypeptide comprising EPO (erythropoietin) and immunoglobulin hybrid Fc, and dosage regimen
Brief description
The present invention relates to a method for treating anemia using a long-acting EPO preparation and, more specifically, to a method for treating an anemia patient through verification of the safe and long-acting optimum effective dosage and usage when a fusion polypeptide containing EPO and immunoglobulin hybrid Fc is administered to the anemia patient. The administration of the fusion polypeptide leads to excellent long-lasting power compared with existing EPO products, and can be effectively used in the treatment of the anemia patient through the dosage and usage suitable to minimize side effects of the cardiovascular system, which may be caused by a rapid elevation reaction of hemoglobin as an anemia treatment effect.
Representative patent
WO2016111575
Category
Method of treatment, Dosage Regimen
Patent holder
GENEXINE, INC. GREEN CROSS CORPORATION
Exclusivity
Not provided
Expiration date
January 8, 2036
Status
Granted: US, KR, RU Pending: BR, CN, ID, JP Not in force: EP
Description
Human IL-1 receptor antagonist and hybrid Fc fusion protein
Brief description
he present invention relates to a fusion protein resulting from the coupling of a human interleukin-1 receptor antagonist and hybrid Fc. More specifically, provided is a fusion protein in which a human interleukin-1 receptor antagonist is coupled to a human immunoglobulin (Ig) hybrid Fc fragment. The hybrid Fc fragment comprises IgD and IgG4. Also provided is a pharmaceutical composition comprising the fusion protein. The pharmaceutical composition of the present invention can be used for the treatment of autoimmune diseases including rheumatoid arthritis, inflammatory bowel disease (e.g. Crohn's disease and ulcerative bowel disease), psoriasis, diabetes and the like. The fusion protein of the present invention can be expected to be usable in the development of new autoimmune-disease drugs
Representative patent
WO2012053828
Category
Compound
Patent holder
Genexine Co Ltd Handok Pharmaceuticals Co Ltd
Exclusivity
Not provided
Expiration date
October 19, 2031
Status
Granted: US, KR
Description
Hybrid human Fc (hyFc) and immunoglobulin fusion protein
Brief description
Disclosed are fusion proteins comprising a biologically active molecule and an immunoglobulin (Ig) Fc domain which is linked to the biologically active molecule. The Fc domain is a hybrid human Fc domain of (i) IgG1, IgG2 or IgG4 or (ii) IgG4 and IgD. The hybrid Fc is useful as a carrier of biologically active molecules.
Representative patent
WO2008147143
Category
Compound
Patent holder
Genexine Co., Ltd.
Exclusivity
Not provided
Expiration date
May 30, 2028
Status
Granted: AU, CA, IL, SG, KR, EP (CH, DE, ES, FR, GB, IT, LI, NL) Pending: BR, JP, CN, HK, RU Not in force: IN
Supporting material
Publications
<p><span style="color: rgb(33, 33, 33);">Kim, Y. J., Koh, E. M., Song, C. H., Byun, M. S., Choi, Y. R., Jeon, E. J., Hwang, K., Kim, S. K., Yang, S. I., & Jung, K. J. (2021). Preclinical immunogenicity testing using anti-drug antibody analysis of GX-G3, Fc-fused recombinant human granulocyte colony-stimulating factor, in rat and monkey models. </span><em style="color: rgb(33, 33, 33);">Scientific reports</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">11</em><span style="color: rgb(33, 33, 33);">(1), 12004. </span><a href="https://doi.org/10.1038/s41598-021-91360-7" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1038/s41598-021-91360-7</a></p>
We optimized and validated analytical tools by adopting validation parameters for immunogenicity assessment. Using these validated tools, we analyzed serum samples from rats and monkeys injected subcutaneously with GX-G3 (1, 3 or 10 mg/kg once a week for 4 weeks followed by a 4-week recovery period) to determine immunogenicity response and toxicokinetic parameters with serum concentration of GX-G3. Several rats and monkeys were determined to be positive for anti-GX-G3 antibodies. Moreover, the immunogenicity response of GX-G3 was lower in monkeys than in rats, which was relevant to show less inhibition of toxicokinetic profiles in monkeys, at least 1 mg/kg administered group, compared to rats. These results suggested the establishment and validation for analyzing anti-GX-G3 antibodies and measurement of serum levels of GX-G3 and anti-GX-G3 antibodies, which were related to toxicokinetic profiles. Taken together, this study provides immunogenicity assessment which is closely implicated with a toxicokinetic study of GX-G3 in 4-week repeated administrated toxicological studies.
<p><span style="color: rgb(33, 33, 33);">Choi, Y. J., Hur, S. Y., Kim, T. J., Hong, S. R., Lee, J. K., Cho, C. H., Park, K. S., Woo, J. W., Sung, Y. C., Suh, Y. S., & Park, J. S. (2020). A Phase II, Prospective, Randomized, Multicenter, Open-Label Study of GX-188E, an HPV DNA Vaccine, in Patients with Cervical Intraepithelial Neoplasia 3. </span><em style="color: rgb(33, 33, 33);">Clinical cancer research : an official journal of the American Association for Cancer Research</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">26</em><span style="color: rgb(33, 33, 33);">(7), 1616–1623. </span><a href="https://doi.org/10.1158/1078-0432.CCR-19-1513" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1158/1078-0432.CCR-19-1513</a></p>
We conducted a prospective, randomized, multicenter, open-label, phase II clinical trial of GX-188E in CIN3 patients positive for human papillomavirus (HPV) type 16/18. The primary endpoint was to determine the histopathologic regression to ≤CIN1 at visit seven (V7; 20 weeks after the first GX-188E injection), and an extension study was pursued until visit 8 (V8; 36 weeks after the first GX-188E injection). HPV-sequencing analysis and an ex vivo IFNγ ELISpot assay were performed using the collected cervical biopsy and blood samples from patients.
In total, 72 patients were enrolled and underwent randomization. Of them, 64 patients were included in per-protocol analysis (V7) and 52 in extension analysis (V8). Our data showed 52% (33/64) of patients at V7 and 67% (35/52) of patients at V8 presented histopathologic regression after receiving the GX-188E injection. We found that 73% (V7) and 77% (V8) of the patients with histologic regression showed HPV clearance.
<p><span style="color: rgb(33, 33, 33);">Youn, J. W., Hur, S. Y., Woo, J. W., Kim, Y. M., Lim, M. C., Park, S. Y., Seo, S. S., No, J. H., Kim, B. G., Lee, J. K., Shin, S. J., Kim, K., Chaney, M. F., Choi, Y. J., Suh, Y. S., Park, J. S., & Sung, Y. C. (2020). Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: interim results of a single-arm, phase 2 trial. </span><em style="color: rgb(33, 33, 33);">The Lancet. Oncology</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">21</em><span style="color: rgb(33, 33, 33);">(12), 1653–1660. </span><a href="https://doi.org/10.1016/S1470-2045(20)30486-1" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1016/S1470-2045(20)30486-1</a></p>
In this open-label, single-arm, phase 2 trial, patients with recurrent or advanced, inoperable cervical cancer, who were aged 18 years or older with Eastern Cooperative Oncology Group performance status of 0 or 1 and histologically confirmed recurrent or advanced HPV-positive (HPV-16 or HPV-18) cervical cancer, and who had progressed after available standard-of-care therapy were recruited from seven hospitals in South Korea. Patients received intramuscular 2 mg GX-188E at weeks 1, 2, 4, 7, 13, and 19, with one optional dose at week 46 that was at the investigator's discretion, and intravenous pembrolizumab 200 mg every 3 weeks for up to 2 years or until disease progression. The primary endpoint was the overall response rate within 24 weeks assessed by the investigator using Response Evaluation Criteria in Solid Tumors version 1.1 in patients who received at least 45 days of treatment 45 days of treatment with at least one post-baseline tumour assessment, and this is the report of a planned interim analysis. This trial is registered with ClinicalTrials.gov, NCT03444376.
<p><span style="color: rgb(33, 33, 33);">Yang, S. H., Yang, S. I., & Chung, Y. K. (2012). A long-acting erythropoietin fused with noncytolytic human Fc for the treatment of anemia. </span><em style="color: rgb(33, 33, 33);">Archives of pharmacal research</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">35</em><span style="color: rgb(33, 33, 33);">(5), 757–759. </span><a href="https://doi.org/10.1007/s12272-012-0500-5" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1007/s12272-012-0500-5</a></p>
The Fc fusion technology has been introduced to generate long-acting antagonistic drugs such as Enbrel, Orencia and Amevive. Here, Genexine created a novel noncytolytic hybrid Fc (hyFc) as a carrier of agonistic protein drugs using naturally existing IgD and IgG4 Fcs without any mutation in the hyFc region. The erythropoietin (EPO) fused with hyFc exhibited little binding activity to FcγR and C1q molecules that are main mediators for death of target cells. The EPO-hyFc showed higher in vitro and in vivo bioactivities than EPOIgG1 Fc and highly glycosylated EPO (Aranesp). Phase I clinical trial with EPO-hyFc is currently undergoing in Korea.
Useful links
There are no additional links
Access principles

|
Collaborate for developmentConsider on a case by case basis, collaborating on developing long acting products with potential significant public health impact, especially for low- and middle-income countries (LMICs), utilising the referred to long-acting technology Not provided |

|
Share technical information for match-making assessmentProvide necessary technical information to a potential partner, under confidentiality agreement, to enable preliminary assessment of whether specific medicines of public health importance in LMICs might be compatible with the referred to long-acting technology to achieve a public health benefit Not provided |

|
Work with MPP to expand access in LMICsIn the event that a product using the referred to long-acting technology is successfully developed, the technology IP holder(s) will work with the Medicines Patent Pool towards putting in place the most appropriate strategy for timely and affordable access in low and middle-income countries, including through licensing Not provided |
Comment & Information
Illustrations
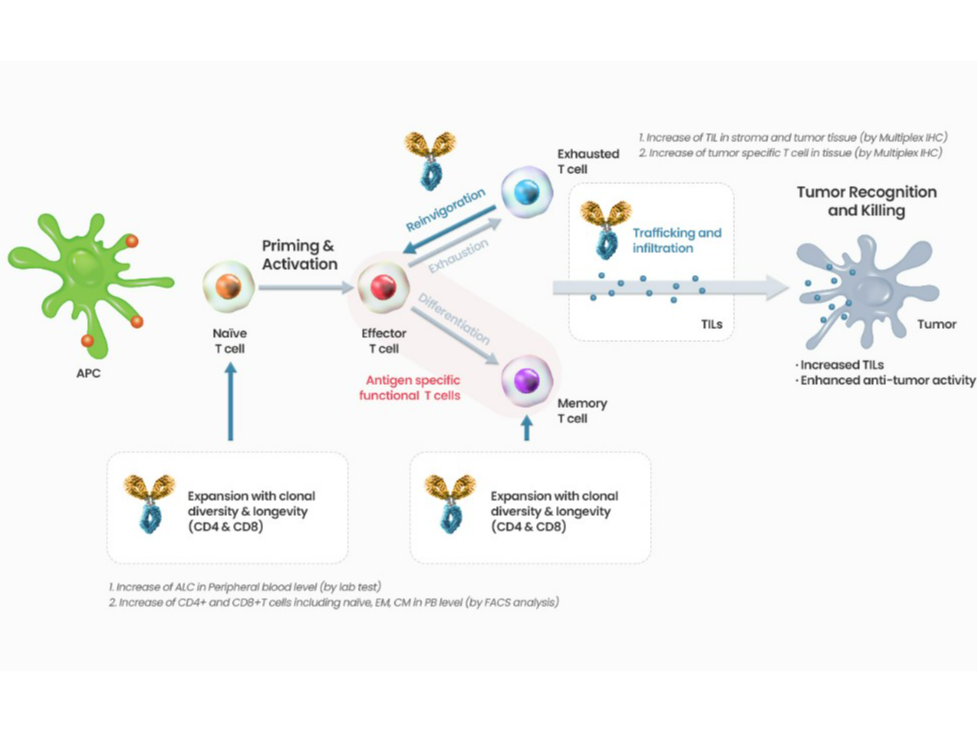
Pharmacodynamics of Gx-i7 inside the cells i.e., hyFc IL-alfa acts on IL-7 receptors, increasing CD4 and CD8 T cells (Naive & memory subtype)
Genexine. (n.d.). GX-I7 pipeline. Genexine. Retrieved September 3, 2024, from http://www.genexine.com/en/pipeline/gx-i7
hyFC engineered IL-7 composed of IgG4 and IgD
Genexine. (n.d.). GX-I7 pipeline. Genexine. Retrieved September 3, 2024, from http://www.genexine.com/en/pipeline/gx-i7
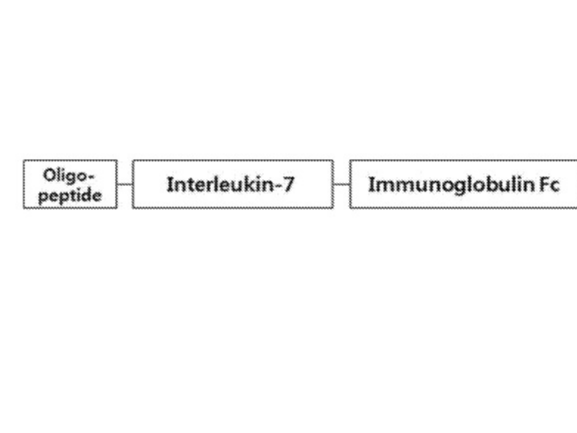
Oligopeptide linkage with IL-7 and IG Fc
U.S. Patent No. US20230279069A1. (2023). Title of the patent. https://patentimages.storage.googleapis.com/00/57/97/83cbd38d28301a/US20230279069A1.pdf
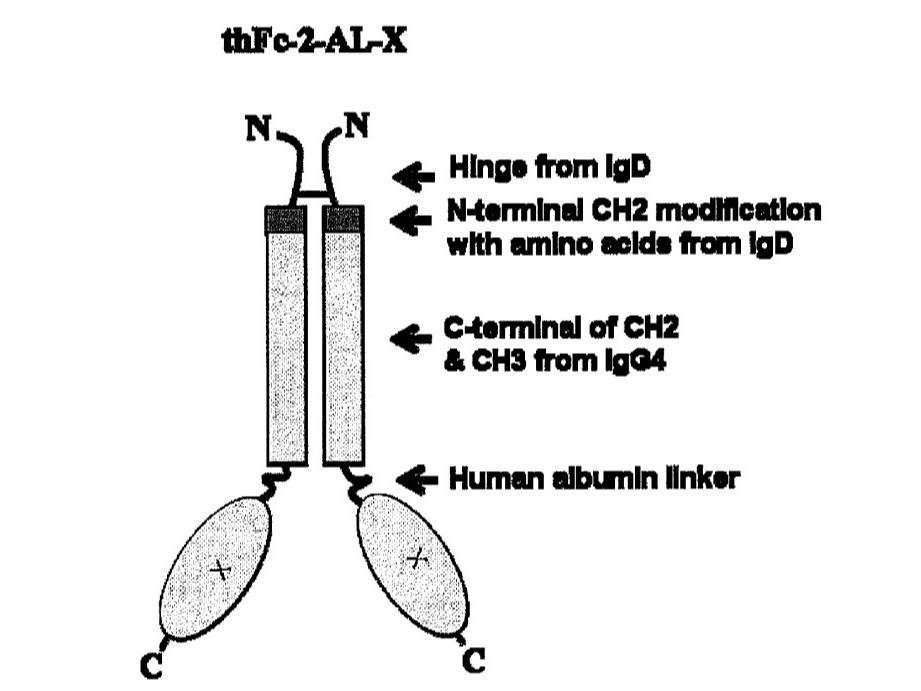
The N terminals of IgD is linked to the compound by a human albumin linker
U.S. Patent No. US7867491B2. (2011). Title of the patent. https://patentimages.storage.googleapis.com/e1/19/d5/2a007ff60f6fe0/US7867491.pdf