|
Developed by 
|
Supported by 
|
TransCon Technology
Developer(s)

|
Ascendis Pharma Originator
https://ascendispharma.com/
Denmark Ascendis Pharma A/S, founded in 2006 in Copenhagen, Denmark, develops innovative therapies using its TransCon® technology, focusing on endocrinology and oncology. With a global presence and ~879 employees, its market cap is ~$7.9B (2025). Recent milestones include YORVIPATH® approval for hypoparathyroidism. Despite revenue challenges with SKYTROFA, it remains committed to advancing its pipeline. |
Sponsor(s)
|
No sponsor indicated |
Partnerships

|
Novo Nordisk https://www.novonordisk.com/ |

|
Teijin, Ltd https://www.teijin.com/ |

|
Pendopharm https://pendopharm.com/ |

|
Adium Pharma https://adiumpharma.com/en/ |

|
Acino https://acino.swiss/ |

|
Vector Pharma, Ltd https://vectorpharma.me/ |

|
SRS Life Sciences https://www.srslife.com/ |

|
VISEN Pharmaceuticals https://www.visenpharma.com/en |

|
Specialised Therapeutics, Inc https://stabiopharma.com/ |
Technology information
Type of technology
Inorganic nanoparticles, Polymer-based particles, Based on other organic particles, Fat based particles, lecithin based particles, Micellar suspension
Administration route
Intravenous, Intratumoral, Subcutaneous
Development state and regulatory approval
Palopegteriparatide
Marketed
YORVIPATH® is approved by the US FDA, EMA, NMPA and Ministry of Food and Drug Safety of South Korea
Description
TransCon technology is a carrier-based system that transiently converts an active drug into a prodrug by conjugating it to a carrier via a cleavable linker. This mechanism enables sustained therapeutic drug levels across systemic and localized administration routes. Initially designed for pediatric sustained-release drug delivery, its application now extends to adult diseases, primarily targeting endocrinological disorders and solid tumors. The choice of carrier and linker is tailored to the route of administration and the desired release kinetics of each API.
Technology highlight
1. TransCon carriers include lipids (ufasomes), polymers (polymerosomes), transfersomes, ethosomes, and sphingosomes. 2. The inertness of the carrier reduces drug clearance, enhancing circulation time. 3. Enables high target-site drug concentrations while minimizing systemic toxicity.
Technology main components
1) API—Can be a small molecule, peptide, or protein. 2) Carrier (with Albumin Avidity)—Either soluble or insoluble, tailored to stability and release profile. 3) Reversible prodrug linker—Aromatic Cyclic Imide, DKP Carbamate, Bicin, AEG (2-((2-aminoethyl)amino)acetic acid), Pyroglutamate 4) Functional agents—antiadsorbents, buffering agents, isotonicity modifiers, preservatives, antimicrobials, stabilizers, diffusion enhancers, and oxidation protectants. 5) Excipients—mannitol, metacresol, sodium hydroxide, succinic acid, water for injection, diluents, adjuvants, and vehicles optimized based on the API’s physicochemical properties.
Information on the raw materials sourcing, availability and anticipated price
Yorvipath 294 micrograms/0.98ml solution for injection prefilled pen (2 pen pack) costs 9272.97 USD
Delivery device(s)
No delivery device
APIs compatibility profile
API desired features
Small molecules
TransCon has the ability to encapsulate small molecules, but the targeted spectrum of pharmacological class is not disclosed.
Nucleic acids
TransCon has the ability to encapsulate RNA molecules, but the targeted spectrum of pharmacological class is not disclosed.
Proteins
Although TransCon has the ability to conjugate with Antibodies, Antibody Fragments, Proteins, Peptides, currently, TransCon is focused on hormones and cell surface proteins targeting neoplastic cells.
Additional solubility data
Not provided
Additional stability data
Not provided
API loading: Maximum drug quantity to be loaded
75-90 wt%
API co-administration
2 different APIs : At least one of the drugs should be a biologically active moiety or a drug that enhances the effect of the other API.
LogP
Not provided
Not provided
Scale-up and manufacturing prospects
Scale-up prospects
Novo Nordisk oversees the commercial manufacturing, clinical development, regulatory approval, and market distribution of products within the TransCon Metabolic Disorders pipeline.
Tentative equipment list for manufacturing
Not provided
Manufacturing
Not provided
Specific analytical instrument required for characterization of formulation
Not provided
Clinical trials
TCP-306
Identifier
NCT05387070
Link
https://clinicaltrials.gov/study/NCT05387070
Phase
Phase III
Status
Recruiting
Sponsor
Visen Pharmaceuticals (Shanghai) Co., Ltd.
More details
This study is limited to conduct in China only. The primary objective is to assess the treatment effect of daily TransCon PTH on serum calcium (sCa) levels within the normal range and stopping from therapeutic doses of active vitamin D (calcitriol) or active vitamin D analogue (alfacalcidol) and calcium at 26 weeks of treatment. All subjects will start with 18 mcg of study drug and will be individually and progressively titrated to an optimal dose over a 26-week double blind period, followed by an open label extension period up to 156 weeks. TransCon PTH or placebo will be administered as a subcutaneous injection using a pre-filled injection pen. Neither trial participants nor their doctors will know who has been assigned to each group. After the 26 weeks, participants will continue in the
Purpose
PaTHway CHINA TRIAL: A Trial to Investigating the Safety, Tolerability and Efficacy of TransCon PTH in Adults With Hypoparathyroidism
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2021-07-28
Anticipated Date of Last Follow-up
2022-05-18
Estimated Primary Completion Date
2022-12-01
Estimated Completion Date
2025-12-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Males and females, ≥18 years of age 2. Subjects with postsurgical chronic HP, or auto-immune, genetic, or idiopathic HP for at least 26 weeks. Diagnosis of HP is established based on historic hypocalcemia in the setting of inappropriately low (below the ULN of local laboratory) serum PTH levels. 3. Requirement for doses of SoC (e.g., calcitriol, alfacalcidol, calcium supplements) at or above a minimum threshold: • requirement for a dose of calcitriol ≥0.5 μg/day, or alfacalcidol ≥1.0 μg/day and (elemental) calcium ≥800 mg/day (e.g., calcium citrate, calcium carbonate etc.) for at least 12 weeks prior to Screening. In addition, the dose of calcitriol, or alfacalcidol, and calcium should be stable for at least 5 weeks prior to Screening 4. Optimization of suppleme
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
76
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
CT301-CN
Identifier
NCT04326374
Link
https://clinicaltrials.gov/study/NCT04326374
Phase
Phase III
Status
Unknown status
Sponsor
Visen Pharmaceuticals (Shanghai) Co., Ltd.
More details
This study is conducted in China only. The purpose is to demonstrate the efficacy and safety of once weekly dosing of TransCon hGH, a long-acting growth hormone product, compare to once-daily dosing of human growth hormone (hGH) after 52 weeks of treatment in prepubertal children with growth hormone deficiency (GHD).
Purpose
Safety, Tolerability and Efficacy of TransCon hGH Weekly Versus Daily hGH in Chinese Pediatric Growth Hormone Deficiency
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2019-12-30
Anticipated Date of Last Follow-up
2020-03-27
Estimated Primary Completion Date
2022-04-01
Estimated Completion Date
2022-04-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Prepubertal children with GHD in Tanner stage 1, aged of 3 years and below 17 years; * Impaired HT defined as at least 2.0 standard deviations (SD) below the mean height for chronological age and sex (HT SDS≤-2.0) according to the Chinese 2005 standard; * Diagnosis of GHD confirmed by 2 different GH stimulation tests, defined as a peak GH level of ≤10 ng/mL, determined with a validated assay. Bone age (BA) at least 6 months less than the chronological age; * Baseline IGF-1 level of at least 1.0 SD below the mean IGF-1 level standardized for age and sex (IGF-1 SDS ≤-1.0); * Written, signed informed consent of the parent(s) or legal guardian(s) of the subject and written assent of the subject (if the subject is 8 years old or above).
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
150
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
enliGHten
Identifier
NCT03344458
Link
https://clinicaltrials.gov/study/NCT03344458
Phase
Phase III
Status
Completed
Sponsor
Ascendis Pharma A/S
More details
A multicenter, phase 3, long-term extension trial of TransCon hGH administered once-weekly in children with growth hormone deficiency (GHD) who previously participated in a phase 3 TransCon hGH trial. Approximately 300 children (males and females) with GHD will be included. All study participants will receive TransCon hGH. This is a global trial that will be conducted in, but not limited to, the United States, Poland, Bulgaria, Ukraine, Armenia, Russia and Australia.
Purpose
A Long-Term Trial Investigating Safety and Efficacy of TransCon hGH in Children With Growth Hormone Deficiency Who Have Completed a Prior TransCon hGH Clinical Trial
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2017-12-19
Anticipated Date of Last Follow-up
2024-04-12
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2023-02-21
Actual Completion Date
2023-02-21
Studied populations
Age Cohort
- Children
- Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Children who have completed a prior phase 3 TransCon hGH trial 2. Children who have not permanently discontinued study drug in the prior trial 3. Written, signed, informed consent of the parent or legal guardian of the subject and written assent of the subject as required by the IRB/HREC/IEC Exclusion Criteria: 1. Poorly-controlled diabetes mellitus (HbA1c ≥ 8.0%) or diabetic complications 2. Evidence of closed epiphyses, defined as bone age \> 14.0 years for females or \> 16.0 years for males 3. Major medical conditions unless approved by Medical Expert 4. Known hypersensitivity to the components of the trial medication 5. Likely to be non-compliant with respect to trial conduct (in regards to the subject and/or the parent/legal guardian/caregiver) 6. Pregnancy
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
298
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TransCon hGH CT-302
Identifier
NCT03305016
Link
https://clinicaltrials.gov/study/NCT03305016
Phase
Phase III
Status
Completed
Sponsor
Ascendis Pharma Endocrinology Division A/S
More details
A 26 week trial of TransCon hGH, a long-acting growth hormone product, administered once-a-week. Approximately 150 children (males and females) with growth hormone deficiency (GHD) will be included. All study participants will receive TransCon hGH. This is a global trial that will be conducted in, but not limited to, the United States, Canada, Australia, and New Zealand.
Purpose
A Safety, Tolerability and Efficacy Study of TransCon hGH in Children With Growth Hormone Deficiency
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2017-11-13
Anticipated Date of Last Follow-up
2021-12-07
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2019-03-19
Actual Completion Date
2019-03-19
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Investigator-determined GHD diagnosis prior to the historical initiation of daily hGH therapy. 2. 6 months to 17 years old, inclusive, at Visit 1 1. If 3 to 17 years old, are taking daily hGH at a dose of ≥ 0.20 mg hGH/kg/week for at least 13 weeks but no more than 130 weeks prior to Visit 1 2. If ≥ 6 months but \< 3 years old, are either hGH treatment-naïve or are taking daily hGH at a dose of ≥ 0.20mg hGH/kg/week for no more than 130 weeks prior to Visit 1 3. Tanner stage \< 5 at Visit 1 4. Open epiphyses (bone age ≤14.0 years for females or ≤16.0 years for males) 5. Written, signed, informed consent of the parent or legal guardian of the subject and written assent of the subject as required by the IRB/HREC/IEC
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
146
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TransCon hGH CT-301
Identifier
NCT02781727
Link
https://clinicaltrials.gov/study/NCT02781727
Phase
Phase III
Status
Completed
Sponsor
Ascendis Pharma Endocrinology Division A/S
More details
A 52 week trial of TransCon hGH, a long-acting growth hormone product, versus human growth hormone therapy. TransCon hGH will be given once-a-week, human growth hormone (hGH) will be given daily. Approximately 150 prepubertal, hGH-treatment naïve children (males and females) with GHD will be included. Randomization will occur in a 2:1 ratio (TransCon hGH : Genotropin). This is a global trial that will be conducted in Armenia, Australia, Belarus, Bulgaria, Georgia, Greece, Italy, New Zealand, Poland, Romania, Russia, Turkey, Ukraine, and the United States.
Purpose
A Phase 3 Trial of the Safety, Tolerability and Efficacy of TransCon hGH Weekly Versus Daily hGH in Children With Growth Hormone Deficiency (GHD)
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2016-12-13
Anticipated Date of Last Follow-up
2021-12-06
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2019-01-17
Actual Completion Date
2019-01-17
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Prepubertal children with GHD (either isolated or as part of a multiple pituitary hormone deficiency) in Tanner stage 1 (Tanner 1982) aged: * Boys: 3-12 years, inclusive * Girls: 3-11 years, inclusive * Impaired height (HT) defined as at least 2.0 standard deviations (SD) below the mean height for chronological age and sex (HT SDS ≤ -2.0) according to the 2000 CDC Growth Charts for the United States Methods and Development, available at http://www.cdc.gov/growthcharts/ * Diagnosis of GHD confirmed by 2 different GH stimulation tests, defined as a peak GH level of ≤10 ng/mL, determined with a validated assay * Bone age (BA) at least 6 months less than chronological age * Baseline IGF-1 level of at least 1 SD below the mean IGF-1 level standardized for age and sex
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
162
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TCH-306EXT
Identifier
NCT05171855
Link
https://clinicaltrials.gov/study/NCT05171855
Phase
Phase III
Status
Completed
Sponsor
Ascendis Pharma Endocrinology Division A/S
More details
This is a phase 3 open-label multicenter extension study designed to evaluate the long-term safety and efficacy of Lonapegsomatropin administered once-weekly. The study participants are adults (males and females) with confirmed growth hormone deficiency (GHD) having completed the treatment period in study TCH-306 (foresiGHt).
Purpose
A Trial to Investigate Long Term Efficacy and Safety of Lonapegsomatropin in Adults With Growth Hormone Deficiency
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2021-12-16
Anticipated Date of Last Follow-up
2025-01-15
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2024-12-23
Actual Completion Date
2024-12-23
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Signing of the trial specific informed consent * Completion of the treatment period and Visit 7 assessments of trial TCH-306, including collection and upload of Visit 7 DXA scan * Fundoscopy at Visit 7 in trial TCH-306 without signs/symptoms of intracranial hypertension or diabetic retinopathy stage 2 / moderate or above Exclusion Criteria: * Diabetes mellitus if any of the following are met: 1. Poorly controlled diabetes, defined as HbA1C higher than 7.5% according to central laboratory at Visit 6 in trial TCH-306 2. Use of diabetes mellitus drugs other than metformin and/or dipeptidyl peptidase-4 (DPP-4) inhibitors * Active malignant disease or history of malignancy.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
233
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
foresiGHt
Identifier
NCT04615273
Link
https://clinicaltrials.gov/study/NCT04615273
Phase
Phase III
Status
Completed
Sponsor
Ascendis Pharma Endocrinology Division A/S
More details
A 38-week dosing trial of lonapegsomatropin, a long-acting growth hormone product, administered once-a-week versus placebo-control. A daily somatropin product arm is also included to assist clinical judgement on the trial results. A total of 264 adults (males and females) with growth hormone deficiency were included. Randomization occurred in a 1:1:1 ratio (lonapegsomatropin: placebo: daily somatropin product). This is a global trial conducted in, but not limited to, the United States, Europe, and Asia.
Purpose
A Trial to Compare the Efficacy and Safety of Once-weekly Lonapegsomatropin With Placebo and a Daily Somatropin Product in Adults With Growth Hormone Deficiency
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2020-12-03
Anticipated Date of Last Follow-up
2024-12-20
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2023-11-02
Actual Completion Date
2023-12-01
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria 1. Age between 23 and 80 years, inclusive, at screening. 2. Adult Growth Hormone Deficiency (AGHD) Diagnosis Criteria * For adult-onset AGHD: documented history of structural hypothalamic-pituitary disease, hypothalamic-pituitary surgery, cranial irradiation, 1-4 non-GH pituitary hormone deficiencies, a proven genetic cause of GHD, or traumatic brain injury (TBI). * Participants with childhood-onset GHD must have had GH axis re-assessed at final height. * In participants with TBI as a cause of GHD, GHD must be confirmed by GH -stimulation testing.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
264
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
ApproaCH
Identifier
NCT05598320
Link
https://clinicaltrials.gov/study/NCT05598320
Phase
Phase II/III
Status
Not provided
Sponsor
Ascendis Pharma Growth Disorders A/S
More details
The purpose of this clinical trial is to evaluate efficacy and safety of once weekly SC doses of 100 µg CNP/kg compared to placebo on Annualized Growth Velocity after a 52-week randomized treatment period in children aged 2 to 11 years with genetically confirmed Achondroplasia. The double-blind, placebo-controlled treatment period is followed by an Open Label Extension (OLE) period of a 52-week duration.
Purpose
A Clinical Trial to Evaluate Efficacy and Safety of TransCon CNP Compared With Placebo in Children With Achondroplasia
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2023-03-03
Anticipated Date of Last Follow-up
2024-10-02
Estimated Primary Completion Date
Not provided
Estimated Completion Date
2025-08-01
Actual Primary Completion Date
2024-08-09
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Written, signed informed consent of the parent(s) or legal guardian(s) of the participant, and as required by the institutional review board/human research ethics committee/independent ethics committee (IRB/HREC/IEC). * Male or female, between 2 and 11 years of age (inclusive) at the time of Screening. * Clinical diagnosis of ACH with documented genetic confirmation available. * Able to stand without assistance. * Parent(s)/legal guardian(s) willing and able to administer weekly SC injections of IMP and to follow the protocol. * At least six months of growth and disease history from ACHieve (TCC-NHS-01) trial or comparable growth and disease history available from medical records (pending confirmation by Medical Monitor). * Considered eligible based on the medical hi
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
84
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
AttaCH
Identifier
NCT05929807
Link
https://clinicaltrials.gov/study/NCT05929807
Phase
Phase II/III
Status
Not provided
Sponsor
Ascendis Pharma Growth Disorders A/S
More details
TransCon CNP administered once-weekly in children and adolescents with achondroplasia who have completed a prior TransCon CNP clinical trial. Participants who complete a prior TransCon CNP trial and meet all eligibility criteria will be invited to continue into the long-term open label extension trial to receive 100 µg CNP/kg/week of TransCon CNP. Trial treatment will be completed when the participant reaches 16 years of age for females and 18 years of age for males and have femur and tibial epiphyseal closure. TransCon CNP treatment will continue if femur and tibial epiphyseal closure is not confirmed at the age of 16 years for females, and 18 years for males. Treatment with TransCon CNP will be completed once femur and tibial epiphyseal closure is confirmed by radiographic imaging. The t
Purpose
A Clinical Trial to Investigate Long-term Safety, Tolerability, and Efficacy of Weekly Subcutaneous Doses With TransCon CNP in Children and Adolescents With Achondroplasia
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2023-06-21
Anticipated Date of Last Follow-up
2024-10-04
Estimated Primary Completion Date
2039-01-01
Estimated Completion Date
2039-03-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Written, signed informed consent of the parent(s) or legal guardian(s) of the participant, and as required by the institutional review board/human research ethics committee/independent ethics committee (IRB/HREC/IEC). For participants who are below the age of consent, a written assent will be obtained in accordance with applicable requirements as required by IRB/HREC/IEC. Upon reaching the legal age of consent, depending on applicable requirements, these participants will be asked to give their own written consent. * Participants with achondroplasia who have completed a clinical trial with TransCon CNP. * Parent(s)/legal guardian(s) willing and able to administer weekly SC injections of TransCon CNP and to follow the protocol. * Considered eligible based on the safet
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
140
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TCC-204
Identifier
NCT05246033
Link
https://clinicaltrials.gov/study/NCT05246033
Phase
Phase II
Status
Not provided
Sponsor
Ascendis Pharma A/S
More details
Purpose of the study: The main purpose of this study is to determine the safety and evaluate the effect of a once weekly dose of TransCon CNP in prepubertal children with achondroplasia in China. Study Treatments: TransCon CNP is an investigational (new) drug, which means that it is currently being tested, and therefore is considered experimental. TransCon CNP is designed to provide a sustained exposure of active CNP by subcutaneous (under the skin) injection once weekly. The Randomized Period of this study is a double-blinded and placebo-controlled. "Placebo-controlled" means that some participants will receive injections that don't contain any TransCon CNP (placebo injection - no active ingredient). "Double-blinded" means that neither the participant nor the study doctor will know wh
Purpose
A Dose Escalation Trial Evaluating Safety, Efficacy, and Pharmacokinetics of Multiple Subcutaneous Doses of TransCon CNP Administered Once Weekly in Children With Achondroplasia
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2022-01-05
Anticipated Date of Last Follow-up
2023-10-31
Estimated Primary Completion Date
Not provided
Estimated Completion Date
2024-03-01
Actual Primary Completion Date
2023-03-15
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Diagnosis of achondroplasia confirmed by genetic testing 2. Age criteria: between ages 2 to 10 years old (inclusive) at Screening Visit 3. Tanner stage 1 breast development for females or testicular volume \< 4ml for males at Screening 4. Able to stand without assistance 5. Parent/ legal guardian willing and able to administer subcutaneous injections of study medication 6. Written, signed informed consent of the parent(s) or legal guardian(s) of the participant and written assent of the participant as required by the institutional review board/human research ethics committee/independent ethics committee (IRB/HREC/IEC).
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
24
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
PaTH Forward
Identifier
NCT04009291
Link
https://clinicaltrials.gov/study/NCT04009291
Phase
Phase II
Status
Not provided
Sponsor
Ascendis Pharma A/S
More details
During the first four weeks of the trial, participants will be randomly assigned to one of four groups: three groups will receive fixed doses of TransCon PTH and one group will receive placebo. TransCon PTH or placebo will be administered as a subcutaneous injection using a pre-filled injection pen. Neither trial participants nor their doctors will know who has been assigned to each group. After the four weeks, participants will continue in the trial as part of a long-term extension study. During the extension, all participants will receive TransCon PTH, with the dose adjusted to their individual needs. This is a global trial that will be conducted in, but not limited to, the United States, Canada, Germany, Denmark, and Norway.
Purpose
A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH in Adults With Hypoparathyroidism
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2019-08-27
Anticipated Date of Last Follow-up
2024-06-13
Estimated Primary Completion Date
Not provided
Estimated Completion Date
2025-03-01
Actual Primary Completion Date
2020-03-06
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Males and females aged ≥18 years. 2. Subjects with postsurgical chronic HP or auto-immune, genetic, or idiopathic HP for at least 26 weeks. 3. On a stable dose for at least 12 weeks (or 4 weeks if on Natpara as of September 2019) prior to Screening of: * ≥0.25 μg BID of calcitriol (active vitamin D) or ≥0.5 μg BID or ≥1.0 μg daily of alfacalcidol (active vitamin D), and * ≥400 mg BID calcium citrate or carbonate. 4. Optimization of supplements prior to randomization to achieve the target levels of: * 25(OH) vitamin D levels of 30-70 ng/mL (75-175 pmol/mL) and * Magnesium level within the normal range and * Albumin-adjusted or ionized serum calcium (sCa) level in the lower half of the normal range. 5. BMI 17-40 kg/m2 at Visit 1. 6. If ≤25 years of ag
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
59
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Not provided
Studied LA-formulation(s)
Not provided
Studied route(s) of administration
Not provided
Use case
Not provided
Key resources
BelieveIT-201
Identifier
NCT05980598
Link
https://clinicaltrials.gov/study/NCT05980598
Phase
Phase II
Status
Not provided
Sponsor
Ascendis Pharma A/S
More details
The purpose of this trial is to evaluate the safety and efficacy of TransCon TLR7/8 Agonist, TransCon IL-2 β/γ, and pembrolizumab given prior to curative intent surgery in treatment of participants with newly diagnosed Stage III/IVA resectable locoregionally advanced head and neck squamous cell carcinoma (LA-HNSCC). After surgery, participants will receive local standard-of-care treatment and will be followed for safety, efficacy, and survival for up to 2 years. This trial contains a safety run-in to evaluate the safety and tolerability of the two treatment arms: Arm A (TransCon TLR7/8 Agonist plus pembrolizumab) and Arm B (TransCon TLR7/8 Agonist plus TransCon IL-2 β/γ). The safety run-in will be followed by the randomized Phase 2, open-label part of the trial comparing the safety, effic
Purpose
TransCon (TC) TLR7/8 Agonist, TC IL-2 β/γ, Pembrolizumab Prior to Surgery for Advanced Head and Neck Squamous Cell Carcinoma
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2023-09-29
Anticipated Date of Last Follow-up
2024-12-02
Estimated Primary Completion Date
2025-04-14
Estimated Completion Date
2027-07-30
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Has local histologically confirmed new diagnosis of resectable, non-metastatic, SCC that is either: Stage III tumor HPV-positive oropharyngeal primary that is tumor size (T) 4, lymph node involvement (N) 0-2, no distant metastases (M) 0; Stage III or IVA oropharyngeal tumor HPV-negative; or Stage III or IVA larynx/hypopharynx/oral cavity primaries regardless of HPV status (per American Joint Committee on Cancer \[AJCC\] Staging, 8th edition). * Has available archived or fresh core or excisional biopsy of a tumor lesion. Note: Fine needle aspirations may be allowed after discussion with Medical Monitor. * Is eligible and plans for primary LA-HNSCC surgery based on investigator decision and per local practice.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
27
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
ASND0030
Identifier
NCT06079398
Link
https://clinicaltrials.gov/study/NCT06079398
Phase
Phase II
Status
Recruiting
Sponsor
Ascendis Pharma A/S
More details
This trial is a Phase 2, multicenter, double-blind, randomized (ratio 2:1 TransCon CNP vs. placebo), placebo-controlled trial, designed to evaluate the safety, tolerability, and efficacy of 100 μg CNP/kg of Navepegritide (TransCon CNP) administered SC once-weekly for 52 weeks in infants with genetically verified heterozygous ACH, aged 0 to \< 2 years at the time of randomization.
Purpose
A Clinical Trial to Evaluate Efficacy and Safety of TransCon CNP Compared With Placebo in Infants (0 to <2 Years of Age) With Achondroplasia
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2024-01-23
Anticipated Date of Last Follow-up
2024-12-23
Estimated Primary Completion Date
2026-03-01
Estimated Completion Date
2027-03-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Children
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Written, signed informed consent by the parent(s)/caregiver(s) of the participant, and as required by the institutional review board/human research ethics committee/independent ethics committee (IRB/HREC/IEC). * Male or female younger than 2 years of age at the time of randomization; or for open label sentinel participants, at the time of first administration of IMP. * Clinical diagnosis of achondroplasia (ACH) with genetic confirmation of heterozygous genotype present during screening. * Parent(s)/caregiver(s) willing to follow the protocol and instructions provided, including being able to administer weekly subcutaneous injections of trial treatment. * Compliance to daily Vitamin D supplementation for infants aged 14 days to 1 year. All participants older than 1 ye
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
72
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Quadruple-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
ACP-001 CT-002
Identifier
NCT01247675
Link
https://clinicaltrials.gov/study/NCT01247675
Phase
Phase II
Status
Completed
Sponsor
Ascendis Pharma A/S
More details
This study investigates the safety, tolerability, pharmacokinetic profile (PK), and pharmacodynamic response (PD) of three different doses of ACP-001 given once-a-week compared to one dose-level of an approved daily human growth hormone product over a period of 4 weeks (4 weekly administrations versus 28 daily administrations) in adults with Growth Hormone Deficiency.
Purpose
A Safety, Pharmacokinetic and Pharmacodynamic Study of ACP-001 (TransCon hGH) in Adults With Growth Hormone Deficiency
Interventions
Intervention 1
Intervention 2
Intervention 3
Intervention 4
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2010-11-01
Anticipated Date of Last Follow-up
2017-01-20
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2011-05-01
Actual Completion Date
2011-05-01
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Male or female between 20 to 70 years * Body Mass Index (BMI, kg/m2) of 19.0 to 36.0 kg/m2, both inclusive * Adult Growth Hormone Deficient (AHGD) patients with documented growth hormone deficiency as defined in the Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II (Consensus Guidelines 1998 and 2007) * Fertile females must agree to use appropriate contraceptive methods and have a negative pregnancy test at inclusion * GH replacement therapy for at least 3 months * Willing to maintain current activity level during the trial * Subjects are able and willing to provide written informed consent and authorization for protected health information disclosure in accordance with Good Clinical Practice (GCP)
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
37
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
IL Believe
Identifier
NCT05081609
Link
https://clinicaltrials.gov/study/NCT05081609
Phase
Phase I/II
Status
Recruiting
Sponsor
Ascendis Pharma Oncology Division A/S
More details
TransCon IL-2 β/γ is an investigational drug being developed for treatment of locally advanced or metastatic solid tumors. This is a first-in-human, open-label, Phase 1/2, dose escalation and dose expansion study of TransCon IL-2 β/γ as monotherapy or in combination therapy in adult participants with advanced or metastatic solid tumors. Given the unique PK profile enabled by the TransCon technology, TransCon IL-2 β/γ presents the opportunity to enhance the therapeutic index of current IL-2 therapy.
Purpose
A Study to Investigate Safety and Tolerability of TransCon IL-2 β/γ Alone or in Combination With Pembrolizumab and/or TransCon TLR7/8 Agonist or Other Anticancer Therapies in Adult Participants With L
Interventions
Intervention 1
Intervention 2
Intervention 3
Intervention 4
Intervention 5
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2022-01-11
Anticipated Date of Last Follow-up
2025-02-03
Estimated Primary Completion Date
2027-08-01
Estimated Completion Date
2029-08-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Key Inclusion Criteria: * At least 18 years of age, or country defined local legal age * Demonstrated adequate organ function at screening * Life expectancy \>12 weeks as determined by the Investigator * Female and male participants of childbearing potential who are sexually active must agree to use highly effective methods of contraception * Participants must have histologically confirmed locally advanced, recurrent, or metastatic solid tumor malignancies that cannot be treated with curative intent (surgery or radiotherapy), with the exception of the neoadjuvant cohorts * Part 1 and Part 2: Eastern Cooperative Oncology Group (ECOG) performance status 0, 1, or 2 * Part 3 and Part 4: Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
345
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TCTLR-101
Identifier
NCT04799054
Link
https://clinicaltrials.gov/study/NCT04799054
Phase
Phase I/II
Status
Not provided
Sponsor
Ascendis Pharma Oncology Division A/S
More details
TransCon TLR7/8 Agonist is an investigational drug being developed for treatment of locally advanced or metastatic solid tumors. This Phase 1/2 study will evaluate TransCon TLR7/8 Agonist as monotherapy or in combination with pembrolizumab in dose escalation and dose expansion. Participants will receive intratumoral (IT) injection of TransCon TLR7/8 Agonist every cycle. The primary objectives are to evaluate safety and tolerability, and define the Maximum Tolerated Dose (MTD) and Recommended Phase 2 Dose (RP2D) of TransCon TLR7/8 Agonist alone or in combination with pembrolizumab.
Purpose
A Study of TransCon TLR7/8 Agonist With or Without Pembrolizumab in Patients With Advanced or Metastatic Solid Tumors
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2021-03-18
Anticipated Date of Last Follow-up
2024-10-29
Estimated Primary Completion Date
2025-05-01
Estimated Completion Date
2026-03-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * At least 18 years of age. * Participants must have histologically confirmed locally advanced, recurrent or metastatic solid tumor malignancies that cannot be treated with curative intent (surgery or radiotherapy). * Participants must have progressed on or be intolerant of available standard of care treatment options or have disease for which there is no standard of care treatment available, with the exception of participants enrolling to the neoadjuvant cohorts. * At least 2 lesions of measurable disease, unless specified otherwise in the selection criteria. * Willingness to undergo biopsies. * Demonstrated adequate organ function within 28 days of Cycle 1 Day 1 (C1D1). * Life expectancy \>12 weeks as determined by the Investigator.
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
188
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TCP-PH-101
Identifier
NCT03803163
Link
https://clinicaltrials.gov/study/NCT03803163
Phase
Phase I
Status
Terminated
Sponsor
Ascendis Pharma A/S
More details
To determine the maximum tolerated dose (MTD) and assess safety and tolerability of escalating single doses of TransCon PEG treprostinil administered as a subcutaneous injection to healthy male volunteers.
Purpose
A Safety, Tolerability and Pharmacokinetics Study of TransCon Treprostinil in Healthy Adult Male Volunteers
Interventions
Not provided
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2015-01-01
Anticipated Date of Last Follow-up
2019-01-11
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2015-06-01
Actual Completion Date
2015-06-01
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: 1. Subject gives voluntary written informed consent to participate in the study. 2. Subject is a healthy male between the ages of 18 and 50 years, inclusive, at Screening. 3. Subjects must weigh between 60 and 120 kg, inclusive, with a BMI between 19.0-32.0 kg/m2, inclusive at Screening. 4. Subject has a medical history, physical examination, vital signs, ECG and clinical laboratory results within normal limits or considered not clinically significant by the Investigator at Screening. 5. Subject agrees to abstain from taking any prescription medication for 14 days prior to check-in and to abstain from taking any non-prescription medications (except multivitamins) or herbal supplements cr
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
20
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
ACP-001 (Prot. 3695)
Identifier
NCT01010425
Link
https://clinicaltrials.gov/study/NCT01010425
Phase
Phase I
Status
Completed
Sponsor
Ascendis Pharma A/S
More details
Double-blind, randomized, placebo and active controlled dose-ascending study to investigate safety, tolerability, pharmacokinetics and pharmacodynamics of ACP-001 (TransCon PEG hGH).
Purpose
Pharmacokinetics and Pharmacodynamics of ACP-001 (TransCon PEG hGH)
Interventions
Intervention 1
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2009-11-01
Anticipated Date of Last Follow-up
2010-06-07
Estimated Primary Completion Date
2014-11-01
Estimated Completion Date
Not provided
Actual Primary Completion Date
2010-02-01
Actual Completion Date
2010-05-01
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: * Healthy male subjects * 20 to 45 years old * Body Mass Index of 18.5 kg/m2 and less than or equal to 29.9 kg/m2 * Others Exclusion Criteria: * Known history of hypersensitivity to human growth hormone (hGH) * Known history of cardiac, pulmonary, gastrointestinal, endocrine, musculoskeletal, neurological, hematological, immunological, hepatic or renal disease, or malignancies * Others
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
44
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
ACP-001 (Prot. 3695)
Identifier
NCT01010425
Link
https://clinicaltrials.gov/study/NCT01010425
Phase
Phase I
Status
Completed
Sponsor
Ascendis Pharma A/S
More details
Double-blind, randomized, placebo and active controlled dose-ascending study to investigate safety, tolerability, pharmacokinetics and pharmacodynamics of ACP-001 (TransCon PEG hGH).
Purpose
Pharmacokinetics and Pharmacodynamics of ACP-001 (TransCon PEG hGH)
Interventions
Intervention 1
Countries
Not provided
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2009-11-01
Anticipated Date of Last Follow-up
2010-06-07
Estimated Primary Completion Date
2014-11-01
Estimated Completion Date
Not provided
Actual Primary Completion Date
2010-02-01
Actual Completion Date
2010-05-01
Studied populations
Age Cohort
- Adults
Genders
- Male
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Yes
Comments about the studied populations
Inclusion Criteria: * Healthy male subjects * 20 to 45 years old * Body Mass Index of 18.5 kg/m2 and less than or equal to 29.9 kg/m2 * Others Exclusion Criteria: * Known history of hypersensitivity to human growth hormone (hGH) * Known history of cardiac, pulmonary, gastrointestinal, endocrine, musculoskeletal, neurological, hematological, immunological, hepatic or renal disease, or malignancies * Others
Health status
Not provided
Study type
Interventional (clinical trial)
Enrollment
44
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Double-blind masking
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
SkyPASS
Identifier
NCT05775523
Link
https://clinicaltrials.gov/study/NCT05775523
Phase
Not provided
Status
Recruiting
Sponsor
Ascendis Pharma Endocrinology Division A/S
More details
The goal of this study is to further characterise the potential long-term safety risks of lonapegsomatropin in patients treated with lonapegsomatropin under real-world conditions in the post-marketing setting.
Purpose
A Post-Authorisation Safety Study (PASS) of Patients Treated With Lonapegsomatropin
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2023-03-20
Anticipated Date of Last Follow-up
2025-01-13
Estimated Primary Completion Date
2033-03-01
Estimated Completion Date
2033-03-01
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
Unspecified
Genders
Unspecified
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
Unspecified
Comments about the studied populations
Not provided
Health status
Not provided
Study type
Not provided
Enrollment
500
Allocation
Not provided
Intervention model
Not provided
Intervention model description
Not provided
Masking
Not provided
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
Excipients
Proprietary excipients used
No proprietary excipient used
Novel excipients or existing excipients at a concentration above Inactive Ingredients Database (IID) for the specified route of administration
No novel excipient or existing excipient used
Residual solvents used
No residual solvent used
Additional features
Other features of the technology
- Biodegradable
- Room temperature storage
- At least 1 year shelf life
Release properties
The unmodified API, encapsulated within the TransCon carrier, is gradually released from the microsphere environment into the surrounding tissues by deconjugating from the linker. This controlled release facilitates the distribution of the API and subsequent activation of target receptors. Additionally, the inactive carrier is dissociated and subsequently eliminated from the system. This mechanism results in pharmacokinetic properties comparable to those observed with daily dosing regimens.
Injectability
TransCon formulations (pre-filled pen) can be administered via intravenous, subcutaneous, or intratumoral routes. These formulations can be injected using a standard injection kit equipped with a 21–25 gauge needle as a ready-to-use liquid formulation with a pen injector, depending on the designated route of administration and the molecular integrity of the formulation.
Safety
In a 52-week efficacy and safety clinical study of TransCon PTH, 72 out of 78 patients (92%) experienced at least one Grade 1 treatment-emergent adverse event (TEAE) by the 52nd week. Additionally, eight patients experienced a serious adverse event (SAE). Treatment-related TEAEs reported in participants included injection site reactions (26%), hypercalcemia (14%), nausea (9%), headache (8%), and hypocalcemia (5%). Two out of 8 SAEs were attributed to hypocalcemia.
Stability
TransCon prefilled pens remain stable for at least one year when stored at cold temperatures.
Storage conditions and cold-chain related features
Do not freeze. Store away from heat. Keep TransCon Formulation in the packaging to protect it from light. Until first use, store the formulation in the refrigerator between 2°C to 8°C. After first use, store it for 14 days at room temperature below 30°C (86°F). After each use, remove the needle and put the pen cap on to protect from light. Discard the prefilled pen 14 days after first use.
Potential application(s)
Therapeutic area(s)
Use case(s)
Use of technology
Ease of administration
- Administered by a community health worker
- Administered by a nurse
- Administered by a specialty health worker
Frequency of administration
Weekly, Monthly
User acceptance
Not provided
Targeted user groups
Age Cohort- Children
- Adolescents
- Adults
- Older Adults
- All
Pregnant individuals
No
Lactating individuals
Unspecified
Healthy individuals
Unspecified
Comment
Not provided
Potential associated API(s)
Human Growth Hormone Agonists, Lonasomatropin
Class(es)
Somatotropins
Development stage
Marketed
Clinical trial number(s)
NCT04326374
Foreseen/approved indication(s)
Pediatric, Adult GHD and Turner Syndrome
Foreseen user group
Children and adults suffering from Growth Hormone Deficiency
Foreseen duration between application(s)
Once weekly
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Lonapegsomatropin-tcgd (SKYTROFA®) was approved by USFDA in August 2021 for treating pediatric GHD. It is also approved by EMA, China, Russia, Belarus and South Korea.
Palopegteriparatide
Class(es)
Somatotropin and somatropin agonists
Development stage
Marketed
Clinical trial number(s)
NCT04009291
Foreseen/approved indication(s)
Pediatric and Adult Hypoparathyroidism
Foreseen user group
Children and adultoscents
Foreseen duration between application(s)
Once weekly
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
YORVIPATH® is approved by the US FDA, EMA, NMPA and Ministry of Food and Drug Safety of South Korea
CNP agonist
Class(es)
Vasodilators
Development stage
Phase II
Clinical trial number(s)
NCT04085523
Foreseen/approved indication(s)
Pediatric Achondroplasia
Foreseen user group
Children and adolescents
Foreseen duration between application(s)
Once weekly
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
TLR7/8 agonist
Class(es)
Immunostimulant - Antineoplastic agent
Development stage
Phase II
Clinical trial number(s)
NCT05980598
Foreseen/approved indication(s)
Solid Tumors
Foreseen user group
Not provided
Foreseen duration between application(s)
Once weekly
Applications to Stringent Regulatory Authorities (SRA) / regulatory approvals
Not provided
Patent info
Description
Combination therapy with controlled-release CNP agonists
Brief description
The present invention relates to combinations, or pharmaceutical compositions comprising, a CNP agonist, preferably a controlled-release CNP agonist, and at least one further biologically active moiety or drug, and their use in methods for the treatment or prevention of disorders that benefit from stimulating growth, such as achondroplasia.
Representative patent
AU2023201709B2
Category
formulation
Patent holder
Ascendis Pharma Endocrinology Division AS
Exclusivity
Not provided
Expiration date
September 28, 2037
Status
Active
Description
Cnp prodrugs with carrier attachment at the ring moiety
Brief description
The present invention relates to CNP prodrugs in which the carrier is covalently and reversibly attached to the ring moiety of a CNP moiety, to pharmaceutical compositions comprising such CNP prodrugs, to their uses and to methods of treating diseases that can be treated with the CNP prodrugs of the present invention.
Representative patent
AU2023210599B2
Category
Not provided
Patent holder
Ascendis Pharma Endocrinology Division AS
Exclusivity
Not provided
Expiration date
May 1, 2037
Status
Active
Supporting material
Publications
<p><span style="color: rgb(33, 33, 33);">Savarirayan, R., Hoernschemeyer, D. G., Ljungberg, M., Zarate, Y. A., Bacino, C. A., Bober, M. B., Legare, J. M., Högler, W., Quattrin, T., Abuzzahab, M. J., Hofman, P. L., White, K. K., Ma, N. S., Schnabel, D., Sousa, S. B., Mao, M., Smith, A., Chakraborty, M., Giwa, A., Winding, B., … McDonnell, C. (2023). Once-weekly TransCon CNP (navepegritide) in children with achondroplasia (ACcomplisH): a phase 2, multicentre, randomised, double-blind, placebo-controlled, dose-escalation trial. </span><em style="color: rgb(33, 33, 33);">EClinicalMedicine</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">65</em><span style="color: rgb(33, 33, 33);">, 102258. </span><a href="https://doi.org/10.1016/j.eclinm.2023.102258" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1016/j.eclinm.2023.102258</a></p>
Abstract
Background: TransCon CNP (navepegritide) is an investigational prodrug of C-type natriuretic peptide (CNP) designed to allow for continuous CNP exposure with once-weekly dosing. This 52-week phase 2 (ACcomplisH) trial assessed the safety and efficacy of TransCon CNP in children with achondroplasia.
Methods: ACcomplisH is a global, randomised, double-blind, placebo-controlled, dose-escalation trial. Study participants were recruited between June 10, 2020, and September 24, 2021. Eligible participants were prepubertal, aged 2-10 years, with genetically confirmed achondroplasia, and randomised 3:1 to once-weekly subcutaneous injections of TransCon CNP (6, 20, 50, or 100 μg CNP/kg/week) or placebo for 52 weeks. Primary objectives were safety and annualised growth velocity (AGV). ACcomplisH is registered with ClinicalTrials.gov (NCT04085523) and Eudra (CT 2019-002754-22).
Findings: Forty-two participants received TransCon CNP at doses of 6 μg (n = 10; 7 female), 20 μg (n = 11; 3 female), 50 μg (n = 10; 3 female), or 100 μg (n = 11; 6 female) CNP/kg/week, with 15 receiving placebo (5 female). Treatment-emergent adverse events (TEAEs) were mild or moderate with no grade 3/4 events reported. There were 2 serious TEAEs that were assessed as not related to TransCon CNP. Eleven injection site reactions occurred in 8 participants receiving TransCon CNP and no symptomatic hypotension occurred. TransCon CNP demonstrated a dose-dependent improvement in AGV. At 52 weeks, TransCon CNP 100 μg CNP/kg/week significantly improved AGV vs placebo (least squares mean [95% CI] 5.42 [4.74-6.11] vs 4.35 [3.75-4.94] cm/year; p = 0.0218), and improved achondroplasia-specific height SDS from baseline (least squares mean [95% CI] 0.22 [0.02-0·41] vs -0·08 [-0.25 to 0.10]; p = 0.0283). All participants completed the randomised period and continued in the ongoing open-label extension period receiving TransCon CNP 100 μg CNP/kg/week.
Interpretation: This phase 2 trial suggests that TransCon CNP is effective, safe, with low injection site reaction frequency, and may provide a novel, once-weekly treatment option for children with achondroplasia. These results support TransCon CNP at 100 μg CNP/kg/week in the ongoing pivotal trial.
<p><span style="color: rgb(33, 33, 33);">Khan, A. A., Rejnmark, L., Rubin, M., Schwarz, P., Vokes, T., Clarke, B., Ahmed, I., Hofbauer, L., Marcocci, C., Pagotto, U., Palermo, A., Eriksen, E., Brod, M., Markova, D., Smith, A., Pihl, S., Mourya, S., Karpf, D. B., & Shu, A. D. (2022). PaTH Forward: A Randomized, Double-Blind, Placebo-Controlled Phase 2 Trial of TransCon PTH in Adult Hypoparathyroidism. </span><em style="color: rgb(33, 33, 33);">The Journal of clinical endocrinology and metabolism</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">107</em><span style="color: rgb(33, 33, 33);">(1), e372–e385. </span><a href="https://doi.org/10.1210/clinem/dgab577" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1210/clinem/dgab577</a></p>
Abstract
Context: Hypoparathyroidism is characterized by insufficient levels of parathyroid hormone (PTH). TransCon PTH is an investigational long-acting prodrug of PTH(1-34) for the treatment of hypoparathyroidism.
Objective: This work aimed to investigate the safety, tolerability, and efficacy of daily TransCon PTH in adults with hypoparathyroidism.
Methods: This phase 2, randomized, double-blind, placebo-controlled 4-week trial with open-label extension enrolled 59 individuals with hypoparathyroidism. Interventions included TransCon PTH 15, 18, or 21 µg PTH(1-34)/day or placebo for 4 weeks, followed by a 22-week extension during which TransCon PTH dose was titrated (6-60 µg PTH[1-34]/day).
Results: By Week 26, 91% of participants treated with TransCon PTH achieved independence from standard of care (SoC, defined as active vitamin D = 0 μg/day and calcium [Ca] ≤ 500 mg/day). Mean 24-hour urine Ca (uCa) decreased from a baseline mean of 415 mg/24h to 178 mg/24h by Week 26 (n = 44) while normal serum Ca (sCa) was maintained and serum phosphate and serum calcium-phosphate product fell within the normal range. By Week 26, mean scores on the generic 36-Item Short Form Health Survey domains increased from below normal at baseline to within the normal range. The Hypoparathyroidism Patient Experience Scale symptom and impact scores improved through 26 weeks. TransCon PTH was well tolerated with no treatment-related serious or severe adverse events.
Conclusion: TransCon PTH enabled independence from oral active vitamin D and reduced Ca supplements (≤ 500 mg/day) for most participants, achieving normal sCa, serum phosphate, uCa, serum calcium-phosphate product, and demonstrating improved health-related quality of life. These results support TransCon PTH as a potential hormone replacement therapy for adults with hypoparathyroidism.
<p><span style="color: rgb(33, 33, 33);">Holten-Andersen, L., Pihl, S., Rasmussen, C. E., Zettler, J., Maitro, G., Baron, J., Heinig, S., Hoffmann, E., Wegge, T., Krusch, M., Faltinger, F., Killian, S., Sprogoe, K., Karpf, D. B., Breinholt, V. M., & Cleemann, F. (2019). Design and Preclinical Development of TransCon PTH, an Investigational Sustained-Release PTH Replacement Therapy for Hypoparathyroidism. </span><em style="color: rgb(33, 33, 33);">Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research</em><span style="color: rgb(33, 33, 33);">, </span><em style="color: rgb(33, 33, 33);">34</em><span style="color: rgb(33, 33, 33);">(11), 2075–2086. </span><a href="https://doi.org/10.1002/jbmr.3824" rel="noopener noreferrer" target="_blank" style="color: rgb(33, 33, 33);">https://doi.org/10.1002/jbmr.3824</a></p>
Abstract
Hypoparathyroidism (HP) is a condition of parathyroid hormone (PTH) deficiency leading to abnormal calcium and phosphate metabolism. The mainstay of therapy consists of vitamin D and calcium supplements, as well as adjunct Natpara (PTH(1-84)). However, neither therapy optimally controls urinary calcium (uCa) or significantly reduces the incidence of hypercalcemia and hypocalcemia. TransCon PTH, a sustained-release prodrug of PTH(1-34) in development for the treatment of HP, was designed to overcome these limitations. To determine the pharmacokinetics and pharmacodynamics of TransCon PTH, single and repeat s.c. dose studies were performed in rats and monkeys. TransCon PTH demonstrated a half-life of 28 and 34 hours in rats and monkeys, respectively. After repeated dosing, an infusion-like profile of the released PTH, characterized by low peak-to-trough levels, was obtained in both species. In intact rats and monkeys, daily subcutaneous administration of TransCon PTH was associated with increases in serum calcium (sCa) levels and decreases in serum phosphate levels (sP). In monkeys, at a single dose of TransCon PTH that increased sCa levels within the normal range, a concurrent decrease in uCa excretion was observed. In 4-week repeat-dose studies in intact rats and monkeys, uCa excretion was comparable to controls across all dose levels despite increases in sCa levels. Further, in a rat model of HP, TransCon PTH normalized sCa and sP levels 24 hours per day. This was in contrast to only transient trends toward normalization of sCa and sP levels with an up to 6-fold higher molar dose of PTH(1-84). After repeated dosing to HP rats, uCa excretion transiently increased, corresponding to increases in sCa above normal range, but at the end of the treatment period, uCa excretion was generally comparable to sham controls. TransCon PTH was well tolerated and the observed pharmacokinetics and pharmacodynamics were in line with the expected action of physiological replacement of PTH.
Additional documents
No documents were uploaded
Useful links
There are no additional links
Access principles

|
Collaborate for developmentConsider on a case by case basis, collaborating on developing long acting products with potential significant public health impact, especially for low- and middle-income countries (LMICs), utilising the referred to long-acting technology Not provided |

|
Share technical information for match-making assessmentProvide necessary technical information to a potential partner, under confidentiality agreement, to enable preliminary assessment of whether specific medicines of public health importance in LMICs might be compatible with the referred to long-acting technology to achieve a public health benefit Not provided |

|
Work with MPP to expand access in LMICsIn the event that a product using the referred to long-acting technology is successfully developed, the technology IP holder(s) will work with the Medicines Patent Pool towards putting in place the most appropriate strategy for timely and affordable access in low and middle-income countries, including through licensing Not provided |
Comment & Information
Illustrations
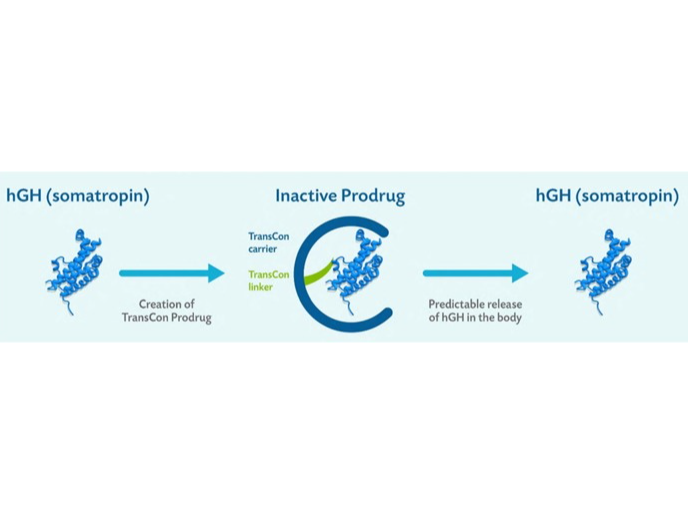
Mechanism of Prodrug conjugation
Sprogøe, K., Mortensen, E., Karpf, D. B., & Leff, J. A. (2017). The rationale and design of TransCon Growth Hormone for the treatment of growth hormone deficiency. Endocrine connections, 6(8), R171–R1
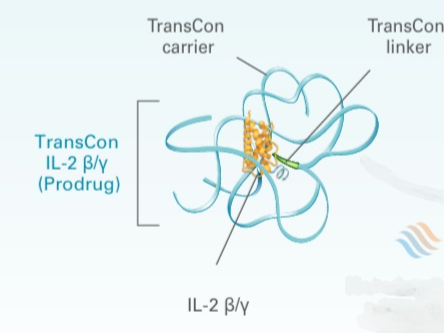
TransCon conjugated complex with IL-2 beta/y linked to the carrier via a Linker
Beyond Achondroplasia. (n.d.). TransCon CNP for achondroplasia. Retrieved February 3, 2025, from https://www.beyondachondroplasia.org/en/health/treatments/emergent-treatments/85-transcon-cnp-for-achon
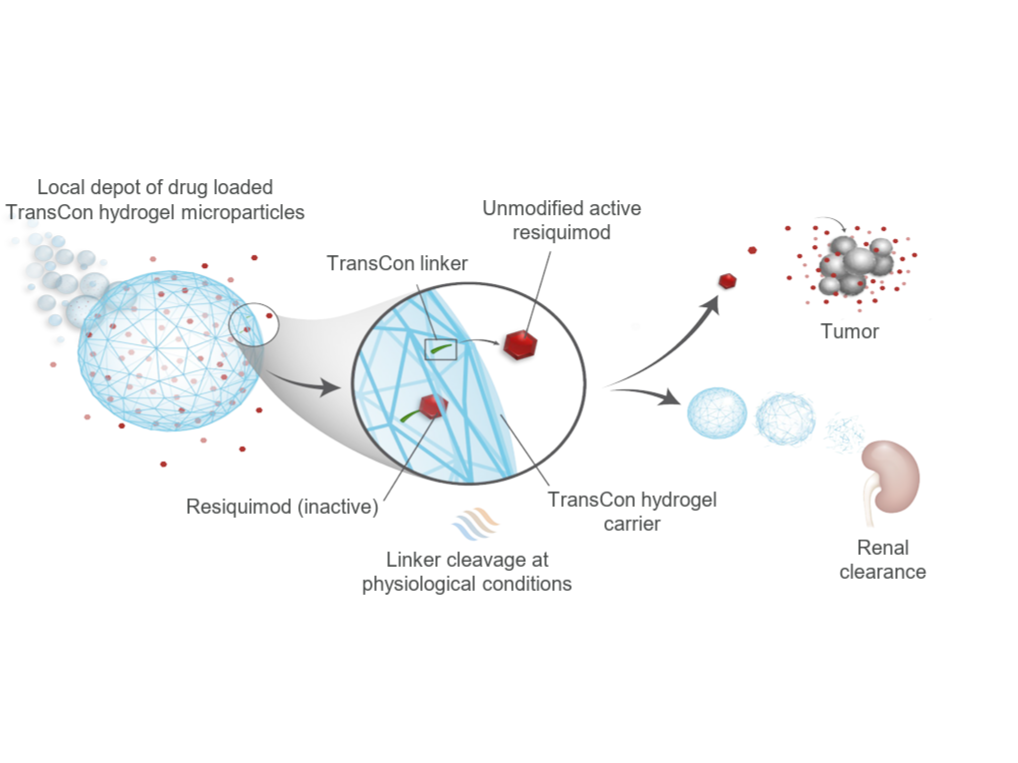
Intracellular deconjugation of TransCon carrier and the linker
Ascendis Pharma. (2025). TransCon TLR7/8 Agonist. Ascendis Pharma. https://investors.ascendispharma.com/static-files/22e32bfb-14ff-4a54-8b47-e49287e24501