Drug name
Last update: Jul 2025Budigalimab
Developer(s)
Drug information
budigalimab
investigational
Biotherapeutic
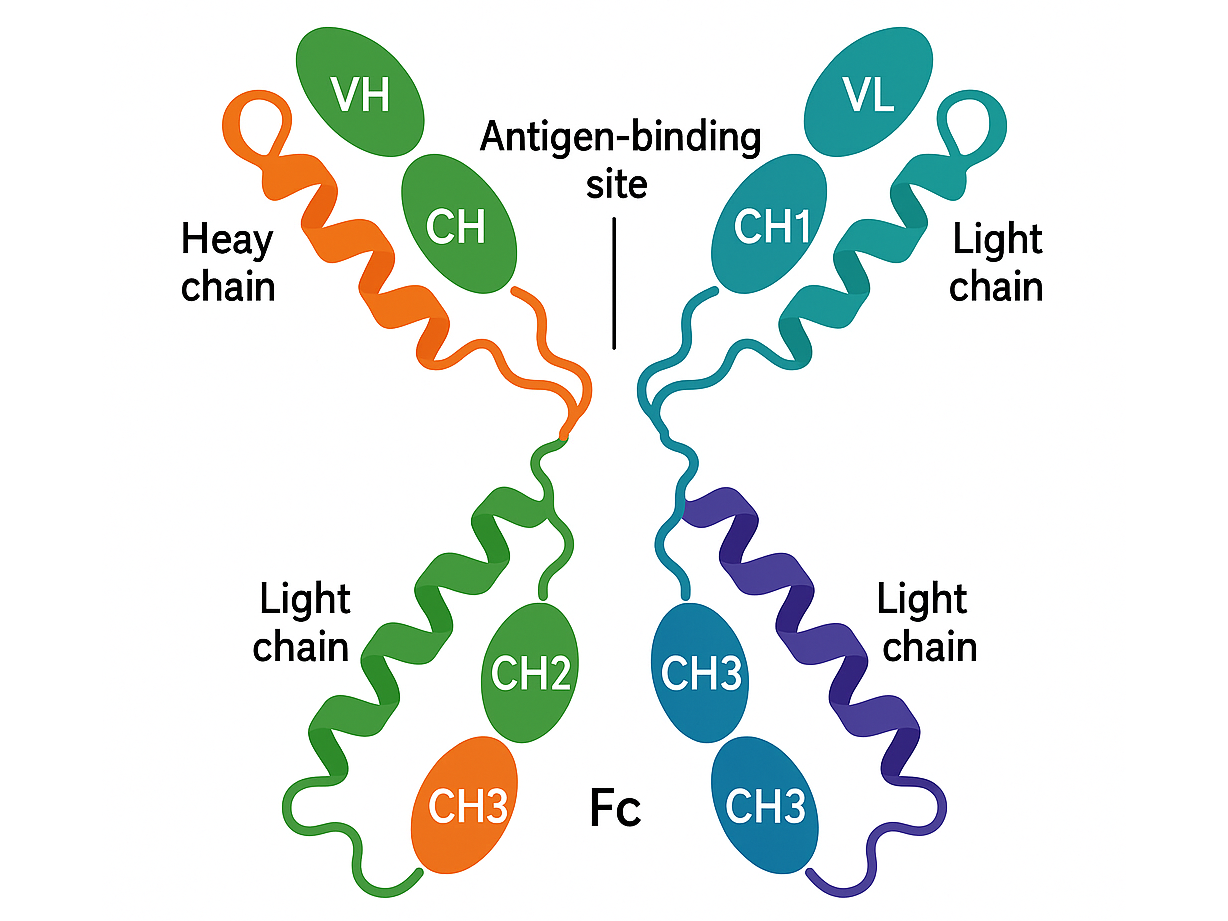
mAb anti-PD1
Budigalimab (ABBV-181) is an investigational anti-PD-1 monoclonal antibody developed for treating solid tumors and hematological malignancies. It binds to the PD-1 receptor on activated T cells, blocking interactions with its ligands, PD-L1 and PD-L2, thereby restoring T-cell activity and promoting anti-tumor immune responses. This mechanism is similar to that of approved checkpoint inhibitors like nivolumab and pembrolizumab. Budigalimab exhibits linear pharmacokinetics, with a half-life of approximately 14–17 days, clearance of 0.22–0.29 L/day, and a volume of distribution of 4–6 L. It reaches steady-state levels after repeated intravenous dosing. Studies show that it also delays HIV rebound or sustained low viral load in a majority of people who interrupted antiretroviral therapy (ART)
Not yet approved
Not approved by any regulatory authorities yet (investigational)
Therapeutic area(s)
- HIV
- Oncology
- Treatment
Administration route
Intravenous
Associated long-acting platforms
Solution
Use of drug
- Administered by a community health worker
- Administered by a nurse
- Administered by a specialty health worker
- Monthly
- Every 2 weeks
Not provided
Dosage
250mg and 500mg
Not available
Budigalimab 250 mg intravenously Q2W Budigalimab 500 mg intravenously Q4W
Not provided
Not provided
Associated technologies
Not provided
Comment & Information
Developer(s)

AbbVie Inc.
AbbVie Inc. is an American pharmaceutical company headquartered in North Chicago, Illinois. It is a global biopharmaceutical company known for its focus on developing treatments in several therapeutic areas, including immunology, oncology, neuroscience, and virology. It was founded in 2013 as a spin-off from Abbott Laboratories to focus specifically on pharmaceuticals.
Drug structure
Scale-up and manufacturing prospects
Not provided
Not provided
• Cell Line Development using CHO (Chinese Hamster Ovary) cell line. • Upstream Processing (USP): Cell Culture & Fermentation in bioreactors • Downstream Processing (DSP): Purification using Ion-Exchange Chromatography • Size exclusion chromatography (SEC) to remove aggregates and fragments
• SEC-HPLC, SDS-PAGE, Mass spectrometry, peptide mapping
Excipients
Not provided
Not provided
Not provided
Delivery device(s)
No delivery device
Method of treatment of HIV using budigalimab and/or ABBV-382
The present disclosure is directed to methods of treating Human Immunodeficiency Virus (HIV) disease, and in particular, to methods of treating HIV disease using anti-PD-1 antibodies and/or anti-α4β7 antibodies, in patients living with HIV who are virologically suppressed on anti-retroviral therapy (ART) undergoing analytical treatment interruption.
WO2025038861
Method of treatment
ABBVIE INC.
Not provided
August 15, 2044
Not yet in national phase , deadline for entry 15 February 2026
Budigalimab compound, including sequences of heavy chain
The present application pertains to, among other things, novel anti-PD-1 antibodies, compositions including the new antibodies, nucleic acids encoding the antibodies, and methods of making and using the same.
WO2018053106
Compound
ABBVIE BIOTHERAPEUTICS INC.
Not provided
September 14, 2037
Granted: CN, CO, EP, HK, JP, KR, MX, ZA, US Pending: AU, BR, CA, VN Withdrawn: CL, CR, DO, EC, ID, GT, MY, PA, PE, PH, RU, UA, TH
Publications
Italiano, A., Cassier, P. A., Lin, C. C., Alanko, T., Peltola, K. J., Gazzah, A., Shiah, H. S., Calvo, E., Cervantes, A., Roda, D., Tosi, D., Gao, B., Millward, M., Warburton, L., Tanner, M., Englert, S., Lambert, S., Parikh, A., Afar, D. E., Vosganian, G., … Moreno, V. (2022). First-in-human phase 1 study of budigalimab, an anti-PD-1 inhibitor, in patients with non-small cell lung cancer and head and neck squamous cell carcinoma. Cancer immunology, immunotherapy : CII, 71(2), 417–431. https://doi.org/10.1007/s00262-021-02973-w
Abstract
Background: Budigalimab is a humanized, recombinant immunoglobulin G1 monoclonal antibody targeting programmed cell death protein 1 (PD-1). We present the safety, efficacy, pharmacokinetic (PK), and pharmacodynamic data from patients enrolled in the head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC) expansion cohorts of the phase 1 first-in-human study of budigalimab monotherapy (NCT03000257; registered 15 December 2016).
Patients and methods: Patients with recurrent/metastatic HNSCC or locally advanced/metastatic NSCLC naive to PD-1/PD-1-ligand inhibitors were enrolled; patients were not selected on the basis of oncogene driver mutations or PD-L1 status. Budigalimab was administered at 250 mg intravenously Q2W or 500 mg intravenously Q4W until disease progression/unacceptable toxicity. The primary endpoints were safety and PK; the secondary endpoint was efficacy. Exploratory endpoints included biomarker assessments.
Results: In total, 81 patients were enrolled (HNSCC: N = 41 [PD-L1 positive: n = 19]; NSCLC: N = 40 [PD-L1 positive: n = 16]); median treatment duration was 72 days (range, 1-617) and 71 days (range, 1-490) for the HNSCC and NSCLC cohorts, respectively. The most frequent grade ≥ 3 treatment-emergent adverse event was anemia (HNSCC: n = 9, 22%; NSCLC: n = 5, 13%). Both dosing regimens had comparable drug exposure and increased interferon gamma-induced chemokines, monokine induced by gamma interferon, and interferon-gamma-inducible protein 10. Objective response rates were 13% (90% CI, 5.1-24.5) in the HNSCC cohort and 19% (90% CI, 9.2-32.6) in the NSCLC cohort. Median progression-free survival was 3.6 months (95% CI, 1.7-4.7) and 1.9 months (95% CI, 1.7-3.7) in the HNSCC and NSCLC cohorts.
Conclusions: The safety, efficacy and biomarker profiles of budigalimab are similar to other PD-1 inhibitors. Development of budigalimab in combination with novel anticancer agents is ongoing.
Calvo, E., Spira, A., Miguel, M., Kondo, S., Gazzah, A., Millward, M., Prenen, H., Rottey, S., Warburton, L., Alanko, T., Cassier, P. A., Yoh, K., Italiano, A., Moreno, V., Peltola, K., Seto, T., Toyozawa, R., Afar, D. E., Englert, S., Komarnitsky, P., … Gao, B. (2021). Safety, pharmacokinetics, and efficacy of budigalimab with rovalpituzumab tesirine in patients with small cell lung cancer. Cancer treatment and research communications, 28, 100405. https://doi.org/10.1016/j.ctarc.2021.100405
Abstract
Background: Agents targeting programmed cell death protein 1 (PD-1) have been approved as monotherapy for patients with small cell lung cancer (SCLC). In preclinical models, the combined targeting of PD-1 and delta-like protein 3 resulted in enhanced antitumor activity. Herein, we report results from the expansion arm of study NCT03000257 evaluating the combination of the anti-PD-1 antibody budigalimab and the targeted antibody-drug conjugate rovalpituzumab tesirine (Rova-T) in patients with previously treated SCLC.
Materials and methods: This expansion arm of a multicenter, open-label, multi-arm, first-in-human phase 1 clinical trial enrolled adult patients with progressive SCLC. The primary objective was to assess safety and tolerability. Patients received budigalimab 375 mg via intravenous infusion every 3 weeks, and Rova-T was administered as a dose of 0.3 mg/kg intravenously, on day 1 of the first and third 3-week cycle.
Results: As of October 2019, 31 patients with SCLC were enrolled and treated with budigalimab plus Rova-T. The combination was tolerated, with the most common treatment-emergent adverse events (in >30%) being pleural effusion, fatigue, and cough. The overall response rate was 24.1%, with one confirmed complete response and six confirmed partial responses. The overall response rate in patients with high delta-like protein 3 expression was similar (21.1%). The median progression-free survival was 3.48 months.
Conclusion: Combination therapy with budigalimab and Rova-T had promising efficacy and appeared to be tolerated in patients with SCLC. Although Rova-T development has been discontinued, development of budigalimab combined with other anticancer agents is ongoing.
Chronic human immunodeficiency virus type 1 (HIV-1) disease results in immune exhaustion and dampened T cell responses, and programmed cell death 1 (PD-1) inhibitors offer a potential approach to enable viral control without antiretroviral therapy (ART) through reversal of these effects. Budigalimab is an investigational humanized anti–PD-1 monoclonal antibody. Multiple intravenous (IV) low doses of budigalimab (Stage 1: 2 mg n = 10, 10 mg n = 10, placebo n = 5, two doses every 4 weeks; Stage 2: 10 mg n = 11, placebo n = 5, four doses every 2 weeks (Q2W)) were assessed in people living with HIV (PLWH; n = 41) in a randomized, double-blind, multicenter, placebo-controlled phase 1 study with an analytical treatment interruption (ATI) to identify an efficacious regimen with a favorable safety profile in PLWH. The primary endpoints were safety, tolerability and pharmacokinetics. Demographics and baseline characteristics were balanced across treatment groups, except for sex, which was mostly male. All participants identified as cisgender. Budigalimab was well tolerated for up to 44 weeks, with 29 of 41 participants experiencing an adverse event (AE), including 2 participants who each experienced one grade 1 reversible immune-related AE (thyroiditis, hyperthyroidism). Three grade 3 AEs were reported by two participants and one serious AE by one participant; none were deemed related to treatment. IV budigalimab 10 mg Q2W resulted in a slight accumulation of drug in serum, with concentrations remaining above the estimated concentration required for near-complete (>95%) PD-1 receptor saturation on CD8+ T cells for ~10 weeks in peripheral blood. In an exploratory efficacy analysis of a 12-week ATI initiated with the first of four 10 mg Q2W doses, 6 of 11 participants experienced a delayed rebound with a relatively low viral peak and/or off-ART viral control (<200 copies ml−1) for ≥6 weeks during ATI, with 2 sustaining ART-free viral control to study end (204−252 days). The study achieved prespecified endpoints, supporting further evaluation of budigalimab in PLWH in a phase 2 study.
Additional documents
No documents were uploaded
Useful links
There are no additional links
Collaborate for development
Consider on a case by case basis, collaborating on developing long acting products with potential significant public health impact, especially for low- and middle-income countries (LMICs), utilising the referred to long-acting technology
Share technical information for match-making assessment
Provide necessary technical information to a potential partner, under confidentiality agreement, to enable preliminary assessment of whether specific medicines of public health importance in LMICs might be compatible with the referred to long-acting technology to achieve a public health benefit
Work with MPP to expand access in LMICs
In the event that a product using the referred to long-acting technology is successfully developed, the technology IP holder(s) will work with the Medicines Patent Pool towards putting in place the most appropriate strategy for timely and affordable access in low and middle-income countries, including through licensing